

Proarrhythmic defects in Timothy syndrome require calmodulin kinase II
- PMID: 19001023
- PMCID: PMC3226825
- DOI: 10.1161/CIRCULATIONAHA.108.788067
Proarrhythmic defects in Timothy syndrome require calmodulin kinase II
Abstract
Background: Timothy syndrome (TS) is a disease of excessive cellular Ca(2+) entry and life-threatening arrhythmias caused by a mutation in the primary cardiac L-type Ca(2+) channel (Ca(V)1.2). The TS mutation causes loss of normal voltage-dependent inactivation of Ca(V)1.2 current (I(Ca)). During cellular Ca(2+) overload, the calmodulin-dependent protein kinase II (CaMKII) causes arrhythmias. We hypothesized that CaMKII is a part of the proarrhythmic mechanism in TS.
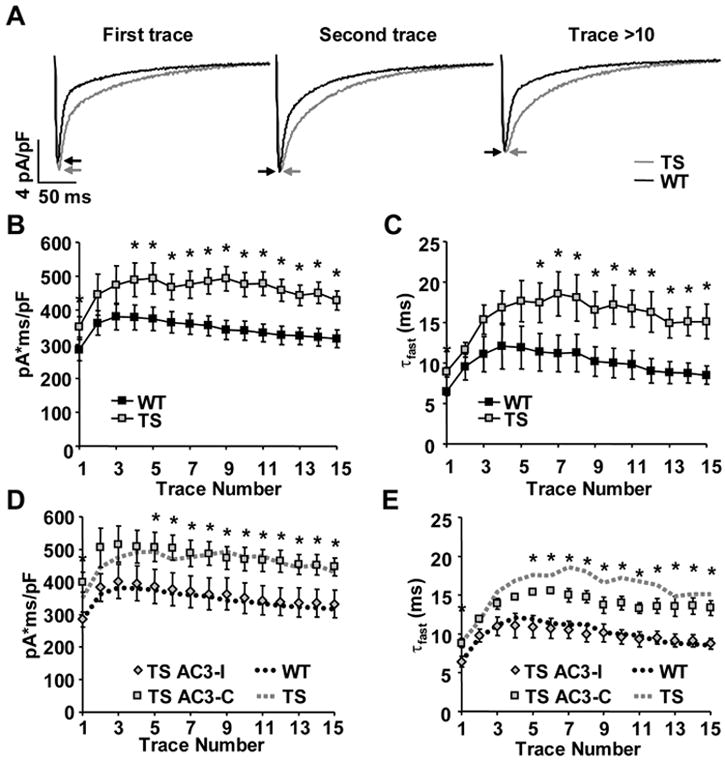
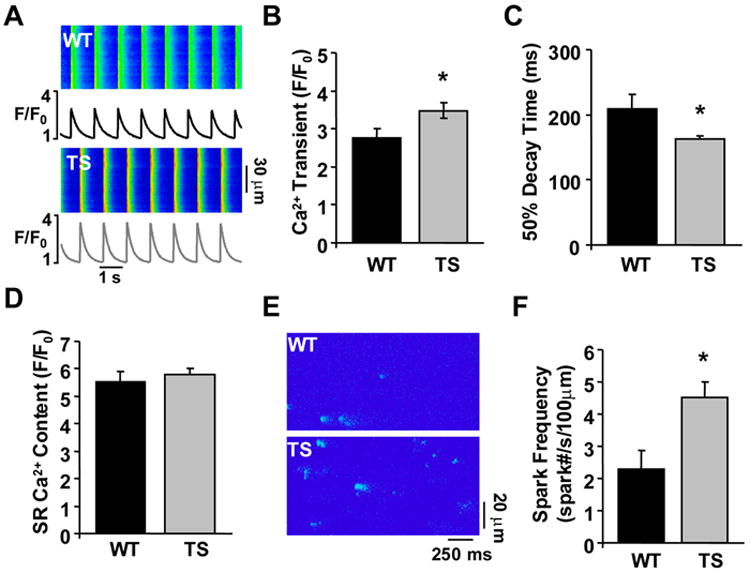
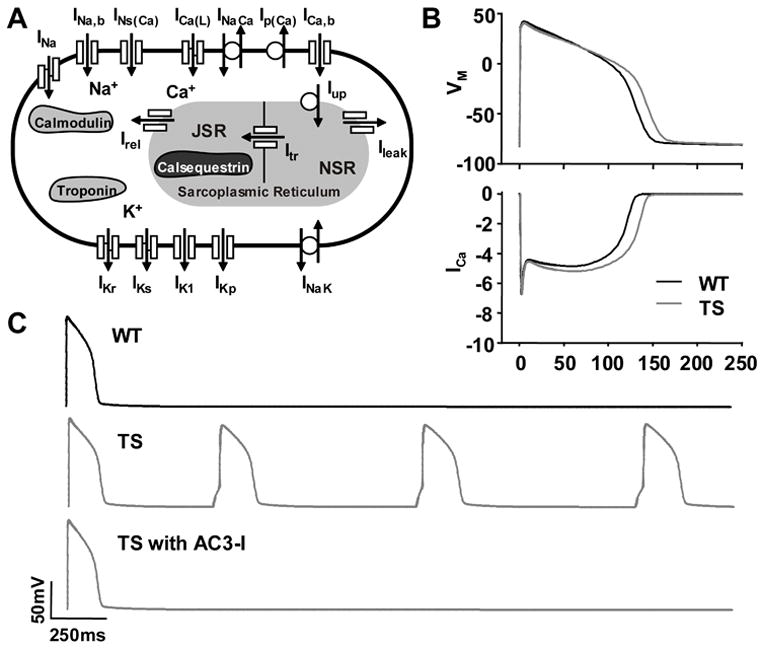
Methods and results: We developed an adult rat ventricular myocyte model of TS (G406R) by lentivirus-mediated transfer of wild-type and TS Ca(V)1.2. The exogenous Ca(V)1.2 contained a mutation (T1066Y) conferring dihydropyridine resistance, so we could silence endogenous Ca(V)1.2 with nifedipine and maintain peak I(Ca) at control levels in infected cells. TS Ca(V)1.2-infected ventricular myocytes exhibited the signature voltage-dependent inactivation loss under Ca(2+) buffering conditions, not permissive for CaMKII activation. In physiological Ca(2+) solutions, TS Ca(V)1.2-expressing ventricular myocytes exhibited increased CaMKII activity and a proarrhythmic phenotype that included action potential prolongation, increased I(Ca) facilitation, and afterdepolarizations. Intracellular dialysis of a CaMKII inhibitory peptide, but not a control peptide, reversed increases in I(Ca) facilitation, normalized the action potential, and prevented afterdepolarizations. We developed a revised mathematical model that accounts for CaMKII-dependent and CaMKII-independent effects of the TS mutation.
Conclusions: In TS, the loss of voltage-dependent inactivation is an upstream initiating event for arrhythmia phenotypes that are ultimately dependent on CaMKII activation.
Conflict of interest statement
Figures
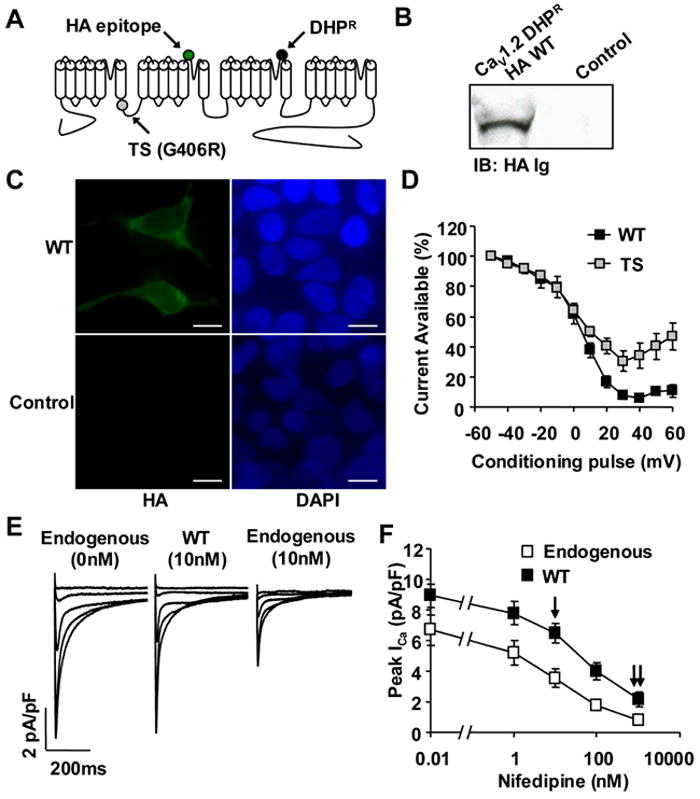
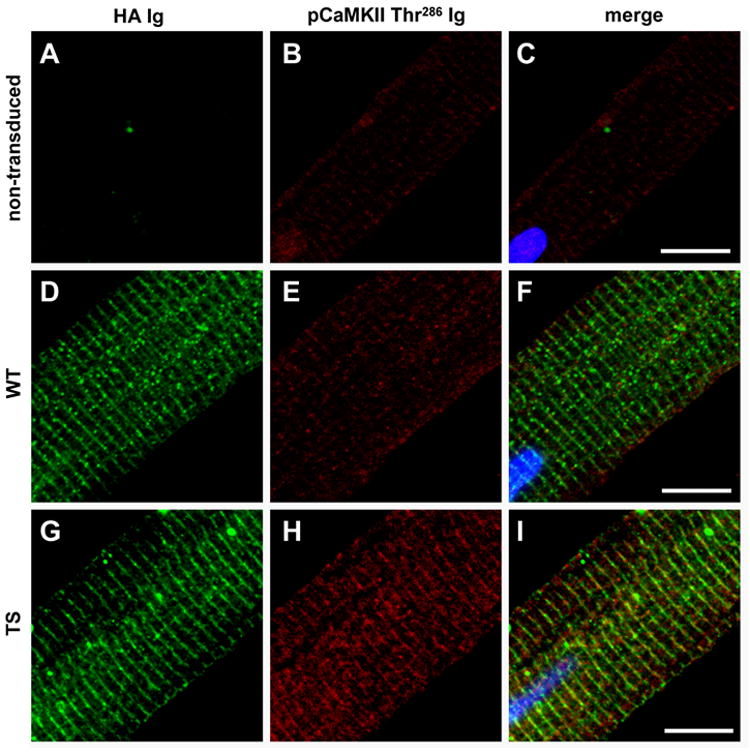
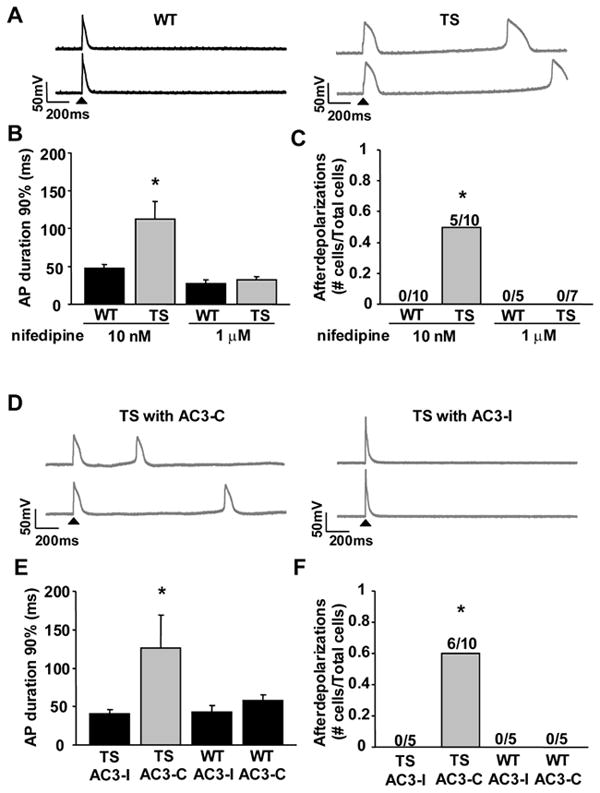
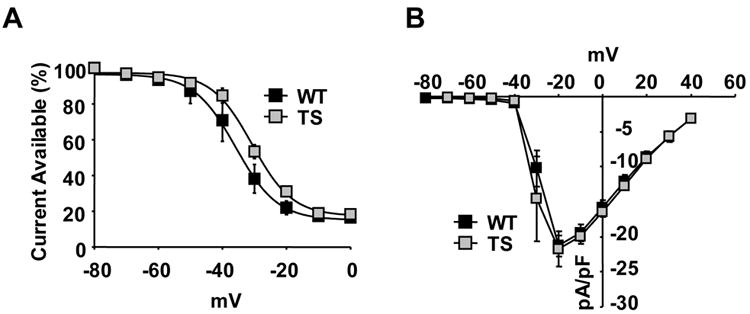
Comment in
-
Understanding cardiac calcium channelopathies.Circulation. 2008 Nov 25;118(22):2221-2. doi: 10.1161/CIRCULATIONAHA.108.819847. Circulation. 2008. PMID: 19029476 No abstract available.
References
-
- Splawski I, Timothy KW, Sharpe LM, Decher N, Kumar P, Bloise R, Napolitano C, Schwartz PJ, Joseph RM, Condouris K, Tager-Flusberg H, Priori SG, Sanguinetti MC, Keating MT. CaV1.2 Calcium Channel Dysfunction Causes a Multisystem Disorder Including Arrhythmia and Autism. Cell. 2004;119:19–31. - PubMed
-
- Zhang R, Khoo MS, Wu Y, Yang Y, Grueter CE, Ni G, Price EE, Thiel W, Guatimosim S, Song LS, Madu EC, Shah AN, Vishnivetskaya TA, Atkinson JB, Gurevich VV, Salama G, Lederer WJ, Colbran RJ, Anderson ME. Calmodulin kinase II inhibition protects against structural heart disease. Nat Med. 2005;11:409–17. - PubMed
-
- Wu Y, Temple J, Zhang R, Dzhura I, Zhang W, Trimble R, Roden DM, Passier R, Olson EN, Colbran RJ, Anderson ME. Calmodulin kinase II and arrhythmias in a mouse model of cardiac hypertrophy. Circulation. 2002;106:1288–93. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
