

Cadherin switching and activation of beta-catenin signaling underlie proinvasive actions of calcitonin-calcitonin receptor axis in prostate cancer
- PMID: 19001380
- PMCID: PMC2613615
- DOI: 10.1074/jbc.M807823200
Cadherin switching and activation of beta-catenin signaling underlie proinvasive actions of calcitonin-calcitonin receptor axis in prostate cancer
Abstract
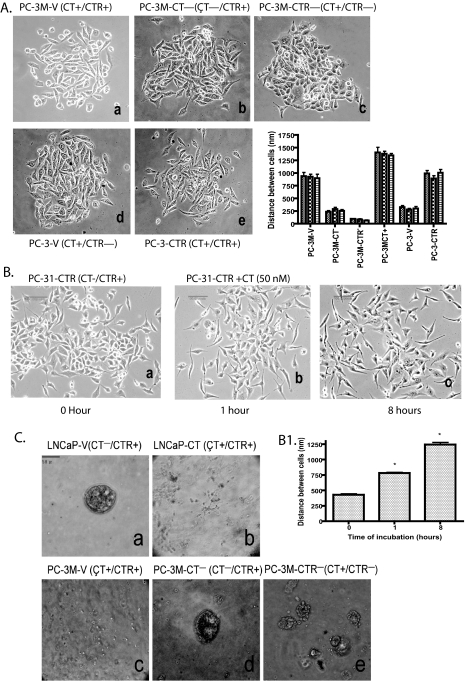
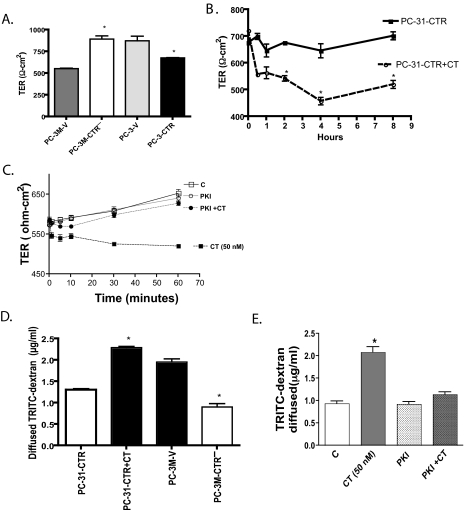
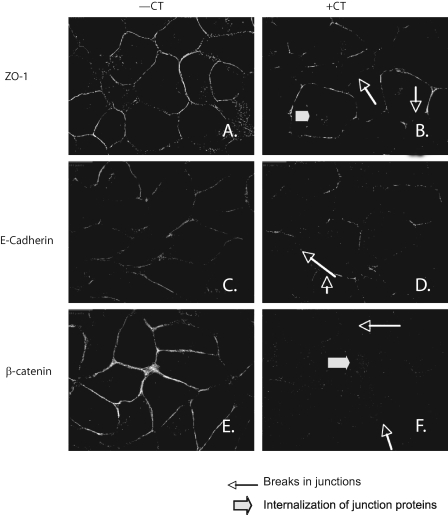
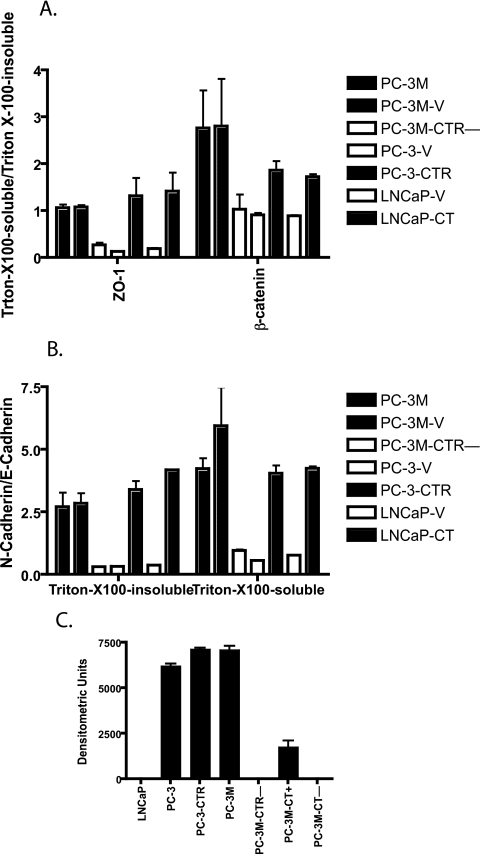
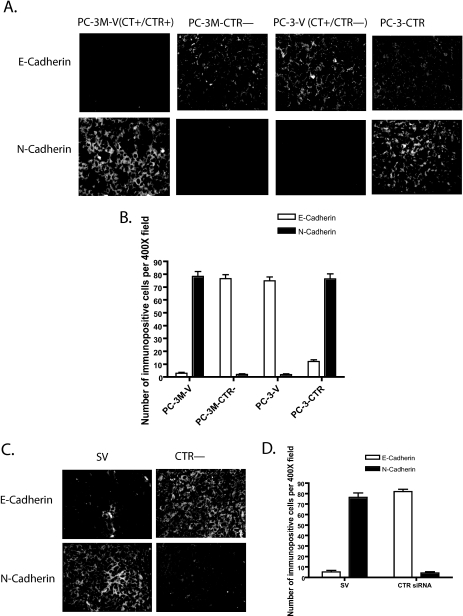
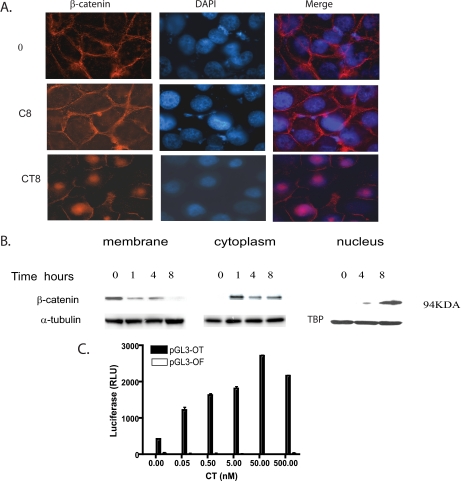
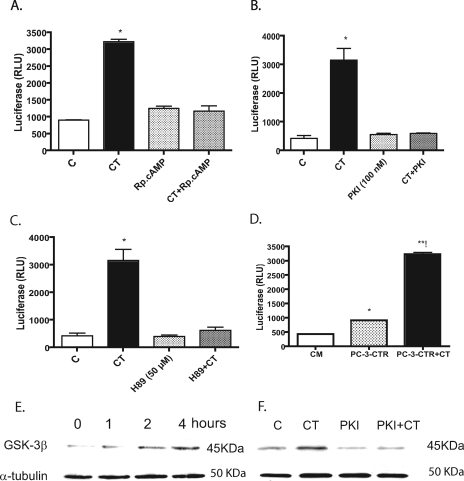
Calcitonin, a neuroendocrine peptide, and its receptor are localized in the basal epithelium of benign prostate but in the secretory epithelium of malignant prostates. The abundance of calcitonin and calcitonin receptor mRNA displays positive correlation with the Gleason grade of primary prostate cancers. Moreover, calcitonin increases tumorigenicity and invasiveness of multiple prostate cancer cell lines by cyclic AMP-dependent protein kinase-mediated actions. These actions include increased secretion of matrix metalloproteinases and urokinase-type plasminogen activator and an increase in prostate cancer cell invasion. Activation of calcitonin-calcitonin receptor autocrine loop in prostate cancer cell lines led to the loss of cell-cell adhesion, destabilization of tight and adherens junctions, and internalization of key integral membrane proteins. In addition, the activation of calcitonin-calcitonin receptor axis induced epithelial-mesenchymal transition of prostate cancer cells as characterized by cadherin switch and the expression of the mesenchymal marker, vimentin. The activated calcitonin receptor phosphorylated glycogen synthase kinase-3, a key regulator of cytosolic beta-catenin degradation within the WNT signaling pathway. This resulted in the accumulation of intracellular beta-catenin, its translocation in the nucleus, and transactivation of beta-catenin-responsive genes. These results for the first time identify actions of calcitonin-calcitonin receptor axis on prostate cancer cells that lead to the destabilization of cell-cell junctions, epithelial-to-mesenchymal transition, and activation of WNT/beta-catenin signaling. The results also suggest that cyclic AMP-dependent protein kinase plays a key role in calcitonin receptor-induced destabilization of cell-cell junctions and activation of WNT-beta-catenin signaling.
Figures

Similar articles
-
Calcitonin receptor increases invasion of prostate cancer cells by recruiting zonula occludens-1 and promoting PKA-mediated TJ disassembly.Cell Signal. 2017 Aug;36:1-13. doi: 10.1016/j.cellsig.2017.04.008. Epub 2017 Apr 17. Cell Signal. 2017. PMID: 28428082
-
Role of Wnt/beta-catenin signaling pathway in epithelial-mesenchymal transition of human prostate cancer induced by hypoxia-inducible factor-1alpha.Int J Urol. 2007 Nov;14(11):1034-9. doi: 10.1111/j.1442-2042.2007.01866.x. Int J Urol. 2007. PMID: 17956532
-
Calcitonin Receptor-Zonula Occludens-1 Interaction Is Critical for Calcitonin-Stimulated Prostate Cancer Metastasis.PLoS One. 2016 Mar 2;11(3):e0150090. doi: 10.1371/journal.pone.0150090. eCollection 2016. PLoS One. 2016. PMID: 26934365 Free PMC article.
-
Beta-catenin and ZO-1: shuttle molecules involved in tumor invasion-associated epithelial-mesenchymal transition processes.Cells Tissues Organs. 2007;185(1-3):61-5. doi: 10.1159/000101304. Cells Tissues Organs. 2007. PMID: 17587809 Review.
-
Roles and regulation of Wnt signaling and beta-catenin in prostate cancer.Cancer Lett. 2006 Jun 8;237(1):22-32. doi: 10.1016/j.canlet.2005.06.004. Epub 2005 Jul 14. Cancer Lett. 2006. PMID: 16023783 Review.
Cited by
-
Epithelial-to-mesenchymal transition in prostate cancer: paradigm or puzzle?Nat Rev Urol. 2011 Jun 21;8(8):428-39. doi: 10.1038/nrurol.2011.85. Nat Rev Urol. 2011. PMID: 21691304 Free PMC article. Review.
-
DDX3X induces primary EGFR-TKI resistance based on intratumor heterogeneity in lung cancer cells harboring EGFR-activating mutations.PLoS One. 2014 Oct 24;9(10):e111019. doi: 10.1371/journal.pone.0111019. eCollection 2014. PLoS One. 2014. PMID: 25343452 Free PMC article.
-
A-kinase anchoring protein 2 is required for calcitonin-mediated invasion of cancer cells.Endocr Relat Cancer. 2016 Jan;23(1):1-14. doi: 10.1530/ERC-15-0425. Epub 2015 Oct 2. Endocr Relat Cancer. 2016. PMID: 26432469 Free PMC article.
-
Relationship between G proteins coupled receptors and tight junctions.Tissue Barriers. 2018 Jan 2;6(1):e1414015. doi: 10.1080/21688370.2017.1414015. Epub 2018 Feb 8. Tissue Barriers. 2018. PMID: 29420165 Free PMC article. Review.
-
Zinc finger protein‑like 1 is a novel neuroendocrine biomarker for prostate cancer.Int J Oncol. 2023 Mar;62(3):38. doi: 10.3892/ijo.2023.5486. Epub 2023 Feb 17. Int J Oncol. 2023. Retraction in: Int J Oncol. 2025 Mar;66(3):16. doi: 10.3892/ijo.2025.5722. PMID: 36799165 Free PMC article. Retracted.
References
-
- Presti, J. C., Jr. (2000) Radiol. Clin. North Am. 38 49-58 - PubMed
-
- Thompson, I. M., Pauler, D. K., Goodman, P. J., Tangen, C. M., Lucia, M. S., Parnes, H. L., Minasian, L. M., Ford, L. G., Lippman, S. M., Crawford, E. D., Crowley, J. J., and Coltman, C. A., Jr. (2004) N. Engl. J. Med. 350 2239-2246 - PubMed
-
- Segawa, N., Mori, I., Utsunomiya, H., Nakamura, M., Nakamura, Y., Shan, L., Kakudo, K., and Katsuoka, Y. (2001) Pathol. Int. 51 452-459 - PubMed
-
- Chien, J., Ren, Y., Qing Wang, Y., Bordelon, W., Thompson, E., Davis, R., Rayford, W., and Shah, G. (2001) Mol. Cell. Endocrinol. 181 69-79 - PubMed
-
- Furuya, Y., Akakura, K., Akimoto, S., Inomiya, H., and Ito, H. (1999) Int. J. Urol. 6 240-244 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
