

Optical lock-in detection imaging microscopy for contrast-enhanced imaging in living cells
- PMID: 19004775
- PMCID: PMC2584753
- DOI: 10.1073/pnas.0808882105
Optical lock-in detection imaging microscopy for contrast-enhanced imaging in living cells
Abstract
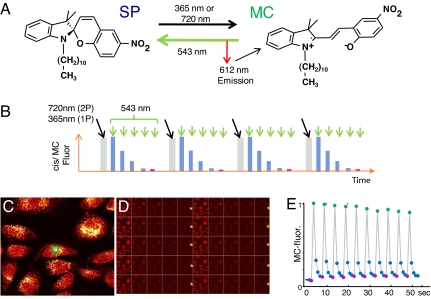
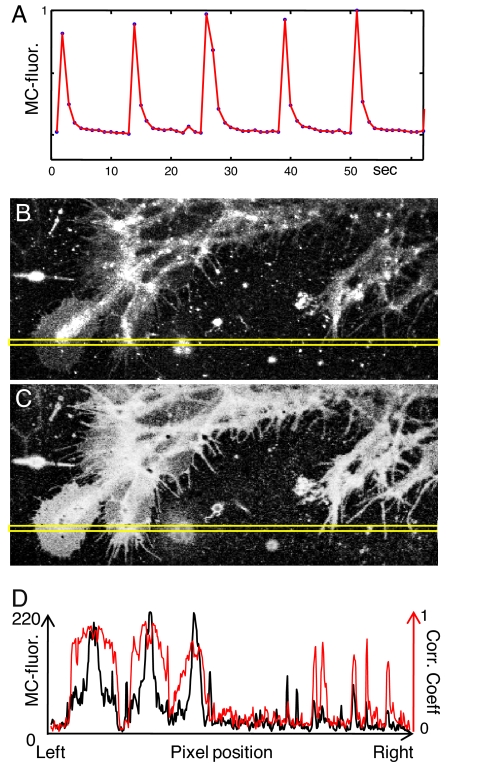
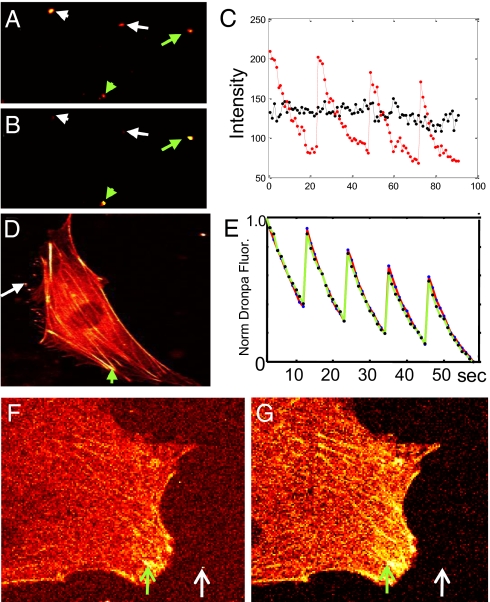
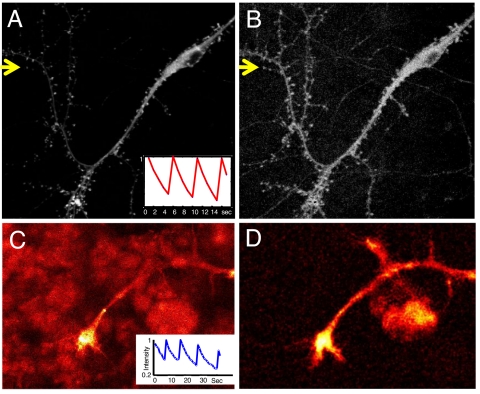
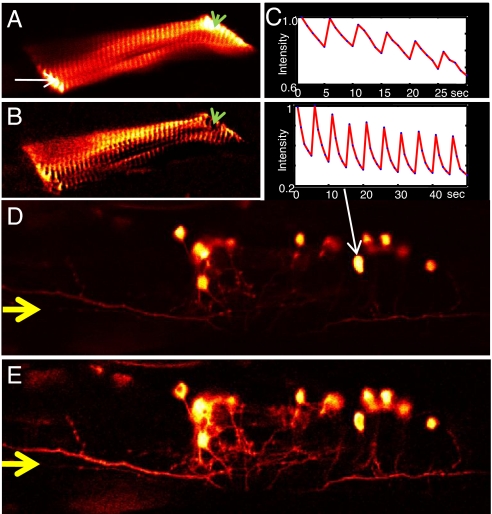
One of the limitations on imaging fluorescent proteins within living cells is that they are usually present in small numbers and need to be detected over a large background. We have developed the means to isolate specific fluorescence signals from background by using lock-in detection of the modulated fluorescence of a class of optical probe termed "optical switches." This optical lock-in detection (OLID) approach involves modulating the fluorescence emission of the probe through deterministic, optical control of its fluorescent and nonfluorescent states, and subsequently applying a lock-in detection method to isolate the modulated signal of interest from nonmodulated background signals. Cross-correlation analysis provides a measure of correlation between the total fluorescence emission within single pixels of an image detected over several cycles of optical switching and a reference waveform detected within the same image over the same switching cycles. This approach to imaging provides a means to selectively detect the emission from optical switch probes among a larger population of conventional fluorescent probes and is compatible with conventional microscopes. OLID using nitrospirobenzopyran-based probes and the genetically encoded Dronpa fluorescent protein are shown to generate high-contrast images of specific structures and proteins in labeled cells in cultured and explanted neurons and in live Xenopus embryos and zebrafish larvae.
Conflict of interest statement
Conflict of interest statement: Related probes to those detailed in this article have been patented by G.M. through the Wisconsin Alumni Research Foundation (WARF).
Figures
References
-
- Yan Y, Marriott G. Analysis of protein interactions using fluorescence technologies. Curr Opin Chem Biol. 2003;7:635–640. - PubMed
-
- Zhang J, Campbell R, Ting A, Tsien R. Creating new fluorescent probes for cell biology. Nat Rev Mol Cell Biol. 2002;3:906–918. - PubMed
-
- Westphal M, et al. Microfilament dynamics in motility and cytokinesis imaged with GFP-actin. Curr Biol. 1997;7:176. - PubMed
-
- Axelrod D. Total internal reflection fluorescence microscopy in cell biology. Methods Enzymol. 2003;361:1–33. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
