

Redox modification of ryanodine receptors contributes to sarcoplasmic reticulum Ca2+ leak in chronic heart failure
- PMID: 19008475
- PMCID: PMC3274754
- DOI: 10.1161/CIRCRESAHA.108.184457
Redox modification of ryanodine receptors contributes to sarcoplasmic reticulum Ca2+ leak in chronic heart failure
Abstract
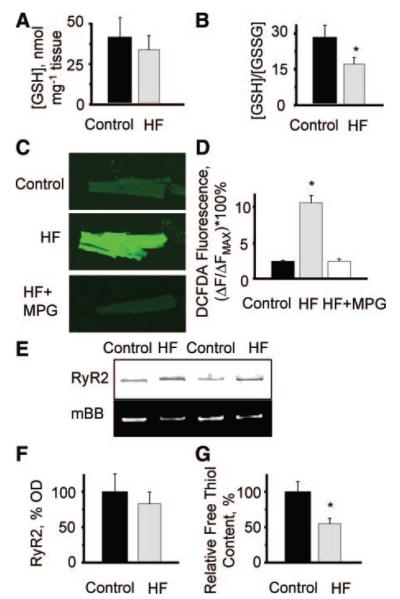
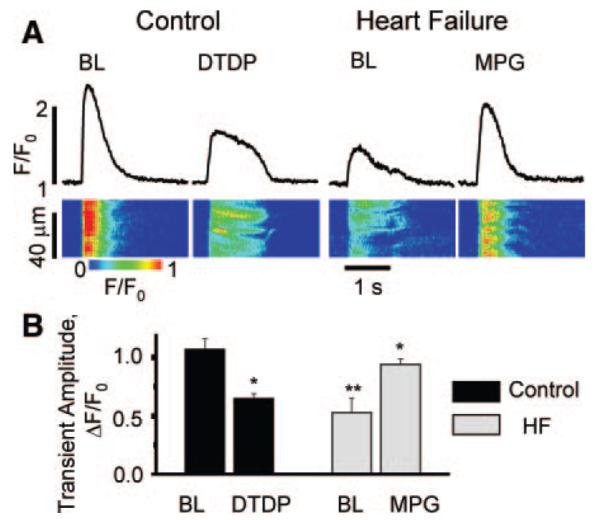
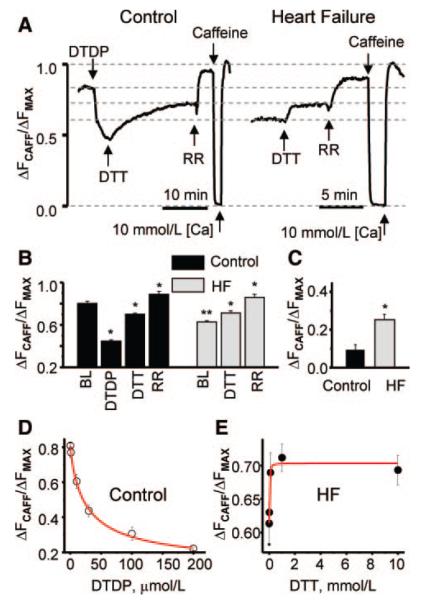
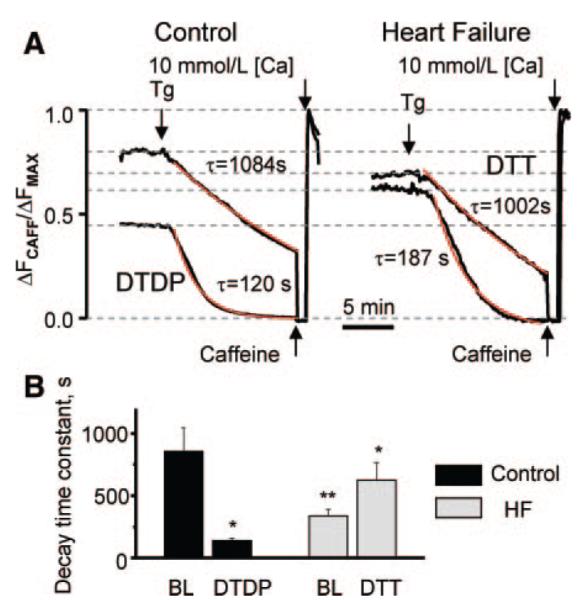
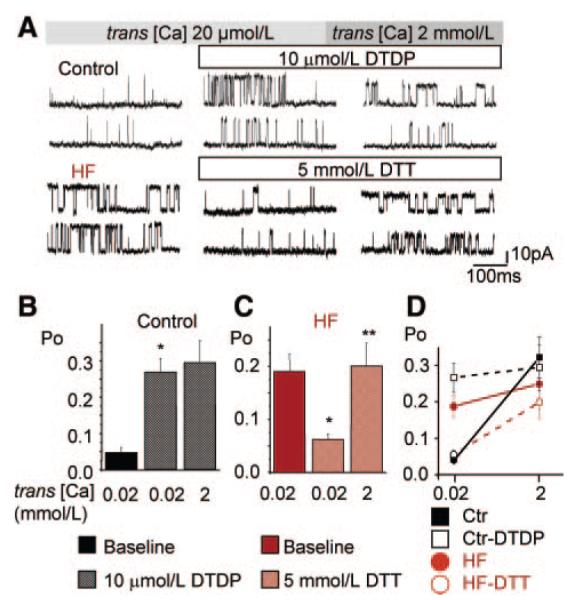
Abnormal cardiac ryanodine receptor (RyR2) function is recognized as an important factor in the pathogenesis of heart failure (HF). However, the specific molecular causes underlying RyR2 defects in HF remain poorly understood. In the present study, we used a canine model of chronic HF to test the hypothesis that the HF-related alterations in RyR2 function are caused by posttranslational modification by reactive oxygen species generated in the failing heart. Experimental approaches included imaging of cytosolic ([Ca(2+)](c)) and sarcoplasmic reticulum (SR) luminal Ca(2+) ([Ca(2+)]SR) in isolated intact and permeabilized ventricular myocytes and single RyR2 channel recording using the planar lipid bilayer technique. The ratio of reduced to oxidized glutathione, as well as the level of free thiols on RyR2 decreased markedly in failing versus control hearts consistent with increased oxidative stress in HF. RyR2-mediated SR Ca(2+) leak was significantly enhanced in permeabilized myocytes, resulting in reduced [Ca(2+)](SR) in HF compared to control cells. Both SR Ca(2+) leak and [Ca(2+)](SR) were partially normalized by treating HF myocytes with reducing agents. Conversely, oxidizing agents accelerated SR Ca(2+) leak and decreased [Ca(2+)](SR) in cells from normal hearts. Moreover, exposure to antioxidants significantly improved intracellular Ca(2+)-handling parameters in intact HF myocytes. Single RyR2 channel activity was significantly higher in HF versus control because of increased sensitivity to activation by luminal Ca(2+) and was partially normalized by reducing agents through restoring luminal Ca(2+) sensitivity oxidation of control RyR2s enhanced their luminal Ca(2+) sensitivity, thus reproducing the HF phenotype. These findings suggest that redox modification contributes to abnormal function of RyR2s in HF, presenting a potential therapeutic target for treating HF.
Figures
References
-
- Lindner M, Erdmann E, Beuckelmann DJ. Calcium content of the sarcoplasmic reticulum in isolated ventricular myocytes from patients with terminal heart failure. J Mol Cell Cardiol. 1998;30:743–749. - PubMed
-
- Hobai IA, O’Rourke B. Decreased sarcoplasmic reticulum calcium content is responsible for defective excitation-contraction coupling in canine heart failure. Circulation. 2001;103:1577–1584. - PubMed
-
- Hasenfuss G, Pieske B. Calcium cycling in congestive heart failure. J Mol Cell Cardiol. 2002;34:951–969. - PubMed
-
- Houser SR, Margulies KB. Is depressed myocyte contractility centrally involved in heart failure? Circ Res. 2003;92:350–358. - PubMed
-
- Sipido KR, Eisner D. Something old, something new: changing views on the cellular mechanisms of heart failure. Cardiovasc Res. 2005;68:167–174. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
