

A predictive model of the oxygen and heme regulatory network in yeast
- PMID: 19008939
- PMCID: PMC2573020
- DOI: 10.1371/journal.pcbi.1000224
A predictive model of the oxygen and heme regulatory network in yeast
Abstract
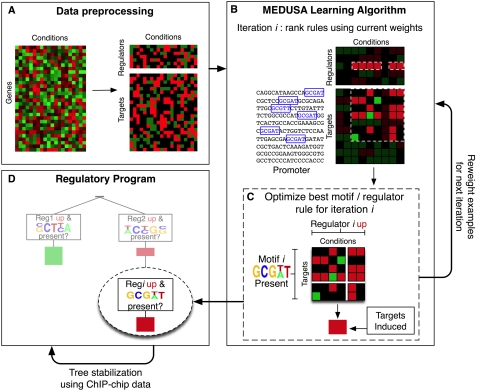
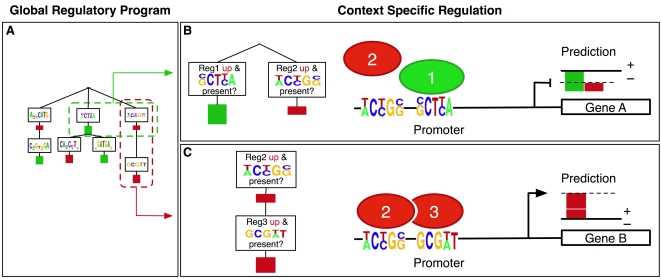
Deciphering gene regulatory mechanisms through the analysis of high-throughput expression data is a challenging computational problem. Previous computational studies have used large expression datasets in order to resolve fine patterns of coexpression, producing clusters or modules of potentially coregulated genes. These methods typically examine promoter sequence information, such as DNA motifs or transcription factor occupancy data, in a separate step after clustering. We needed an alternative and more integrative approach to study the oxygen regulatory network in Saccharomyces cerevisiae using a small dataset of perturbation experiments. Mechanisms of oxygen sensing and regulation underlie many physiological and pathological processes, and only a handful of oxygen regulators have been identified in previous studies. We used a new machine learning algorithm called MEDUSA to uncover detailed information about the oxygen regulatory network using genome-wide expression changes in response to perturbations in the levels of oxygen, heme, Hap1, and Co2+. MEDUSA integrates mRNA expression, promoter sequence, and ChIP-chip occupancy data to learn a model that accurately predicts the differential expression of target genes in held-out data. We used a novel margin-based score to extract significant condition-specific regulators and assemble a global map of the oxygen sensing and regulatory network. This network includes both known oxygen and heme regulators, such as Hap1, Mga2, Hap4, and Upc2, as well as many new candidate regulators. MEDUSA also identified many DNA motifs that are consistent with previous experimentally identified transcription factor binding sites. Because MEDUSA's regulatory program associates regulators to target genes through their promoter sequences, we directly tested the predicted regulators for OLE1, a gene specifically induced under hypoxia, by experimental analysis of the activity of its promoter. In each case, deletion of the candidate regulator resulted in the predicted effect on promoter activity, confirming that several novel regulators identified by MEDUSA are indeed involved in oxygen regulation. MEDUSA can reveal important information from a small dataset and generate testable hypotheses for further experimental analysis. Supplemental data are included.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures







References
-
- Bunn HF, Poyton RO. Oxygen sensing and molecular adaptation to hypoxia. Physiol Rev. 1996;76:839–885. - PubMed
-
- Tai SL, Boer VM, Daran-Lapujade P, Walsh MC, de Winde JH, et al. Two-dimensional transcriptome analysis in chemostat cultures. Combinatorial effects of oxygen availability and macronutrient limitation in Saccharomyces cerevisiae. J Biol Chem. 2005;280:437–447. - PubMed
-
- Piper MD, Daran-Lapujade P, Bro C, Regenberg B, Knudsen S, et al. Reproducibility of oligonucleotide microarray transcriptome analyses. An interlaboratory comparison using chemostat cultures of Saccharomyces cerevisiae. J Biol Chem. 2002;277:37001–37008. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
