

The host cell sulfonation pathway contributes to retroviral infection at a step coincident with provirus establishment
- PMID: 19008949
- PMCID: PMC2576444
- DOI: 10.1371/journal.ppat.1000207
The host cell sulfonation pathway contributes to retroviral infection at a step coincident with provirus establishment
Abstract
The early steps of retrovirus replication leading up to provirus establishment are highly dependent on cellular processes and represent a time when the virus is particularly vulnerable to antivirals and host defense mechanisms. However, the roles played by cellular factors are only partially understood. To identify cellular processes that participate in these critical steps, we employed a high volume screening of insertionally mutagenized somatic cells using a murine leukemia virus (MLV) vector. This approach identified a role for 3'-phosphoadenosine 5'-phosphosulfate synthase 1 (PAPSS1), one of two enzymes that synthesize PAPS, the high energy sulfate donor used in all sulfonation reactions catalyzed by cellular sulfotransferases. The role of the cellular sulfonation pathway was confirmed using chemical inhibitors of PAPS synthases and cellular sulfotransferases. The requirement for sulfonation was mapped to a stage during or shortly after MLV provirus establishment and influenced subsequent gene expression from the viral long terminal repeat (LTR) promoter. Infection of cells by an HIV vector was also shown to be highly dependent on the cellular sulfonation pathway. These studies have uncovered a heretofore unknown regulatory step of retroviral replication, have defined a new biological function for sulfonation in nuclear gene expression, and provide a potentially valuable new target for HIV/AIDS therapy.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
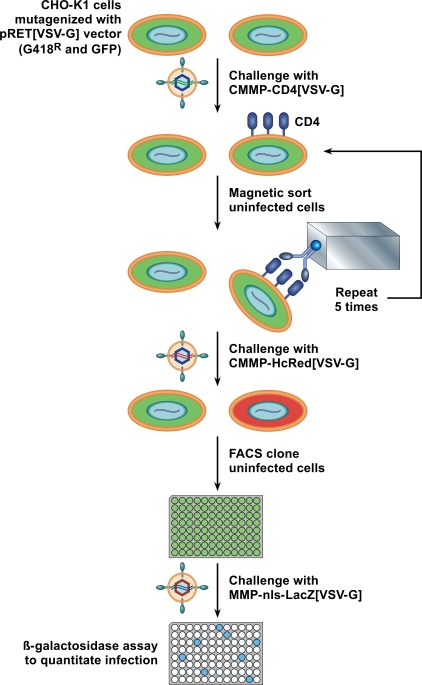



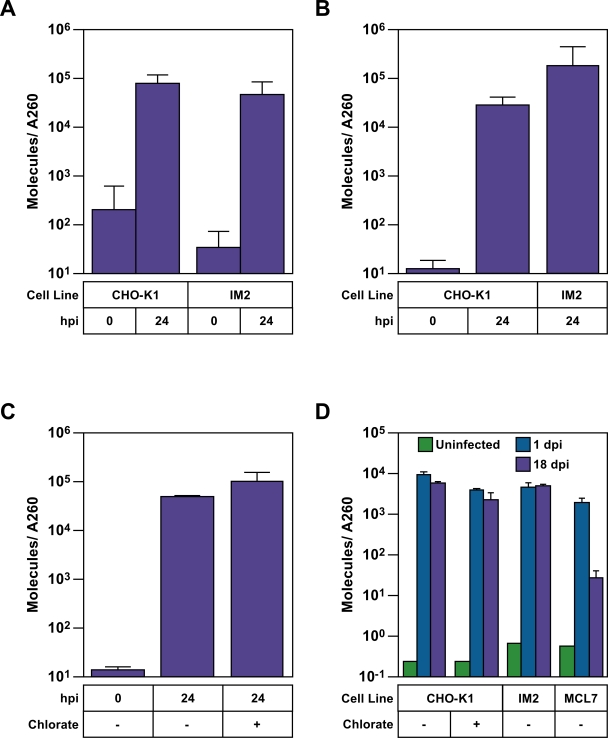



References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
