

Genetic predictors of MEK dependence in non-small cell lung cancer
- PMID: 19010912
- PMCID: PMC2649746
- DOI: 10.1158/0008-5472.CAN-08-2223
Genetic predictors of MEK dependence in non-small cell lung cancer
Abstract
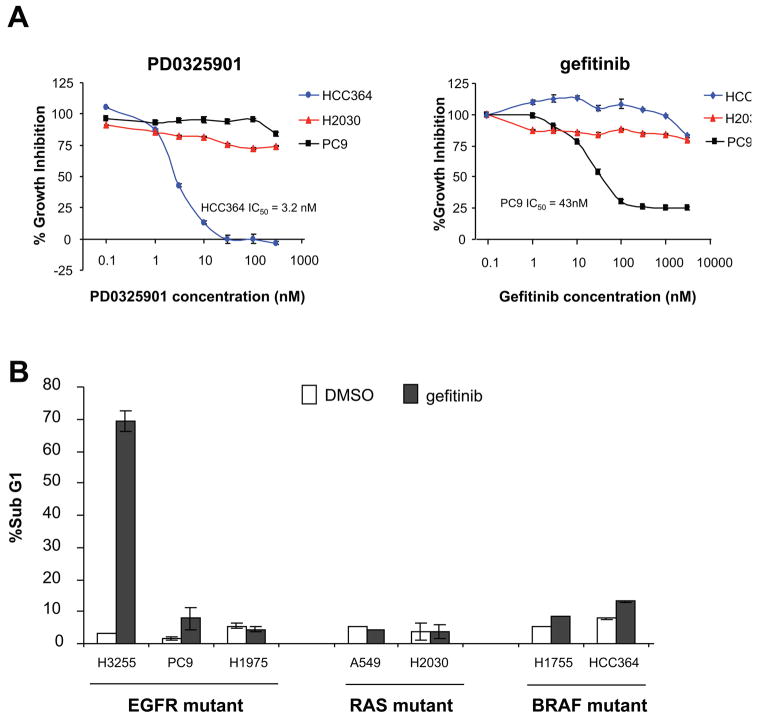
Hyperactivated extracellular signal-regulated kinase (ERK) signaling is common in human cancer and is often the result of activating mutations in BRAF, RAS, and upstream receptor tyrosine kinases. To characterize the mitogen-activated protein kinase/ERK kinase (MEK)/ERK dependence of lung cancers harboring BRAF kinase domain mutations, we screened a large panel of human lung cancer cell lines (n = 87) and tumors (n = 916) for BRAF mutations. We found that non-small cell lung cancers (NSCLC) cells with both V600E and non-V600E BRAF mutations were selectively sensitive to MEK inhibition compared with those harboring mutations in epidermal growth factor receptor (EGFR), KRAS, or ALK and ROS kinase fusions. Supporting its classification as a "driver" mutation in the cells in which it is expressed, MEK inhibition in (V600E)BRAF NSCLC cells led to substantial induction of apoptosis, comparable with that seen with EGFR kinase inhibition in EGFR mutant NSCLC models. Despite high basal ERK phosphorylation, EGFR mutant cells were uniformly resistant to MEK inhibition. Conversely, BRAF mutant cell lines were resistant to EGFR inhibition. These data, together with the nonoverlapping pattern of EGFR and BRAF mutations in human lung cancer, suggest that these lesions define distinct clinical entities whose treatment should be guided by prospective real-time genotyping. To facilitate such an effort, we developed a mass spectrometry-based genotyping method for the detection of hotspot mutations in BRAF, KRAS, and EGFR. Using this assay, we confirmed that BRAF mutations can be identified in a minority of NSCLC tumors and that patients whose tumors harbor BRAF mutations have a distinct clinical profile compared with those whose tumors harbor kinase domain mutations in EGFR.
Figures




References
-
- American Cancer Society. Cancer Facts and Figures. Atlanta: American Cancer Society; 2008.
-
- Rikova K, Guo A, Zeng Q, et al. Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell. 2007;131:1190–203. - PubMed
-
- Lynch TJ, Bell DW, Sordella R, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129–39. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
