

Single-RNA counting reveals alternative modes of gene expression in yeast
- PMID: 19011635
- PMCID: PMC3154325
- DOI: 10.1038/nsmb.1514
Single-RNA counting reveals alternative modes of gene expression in yeast
Abstract
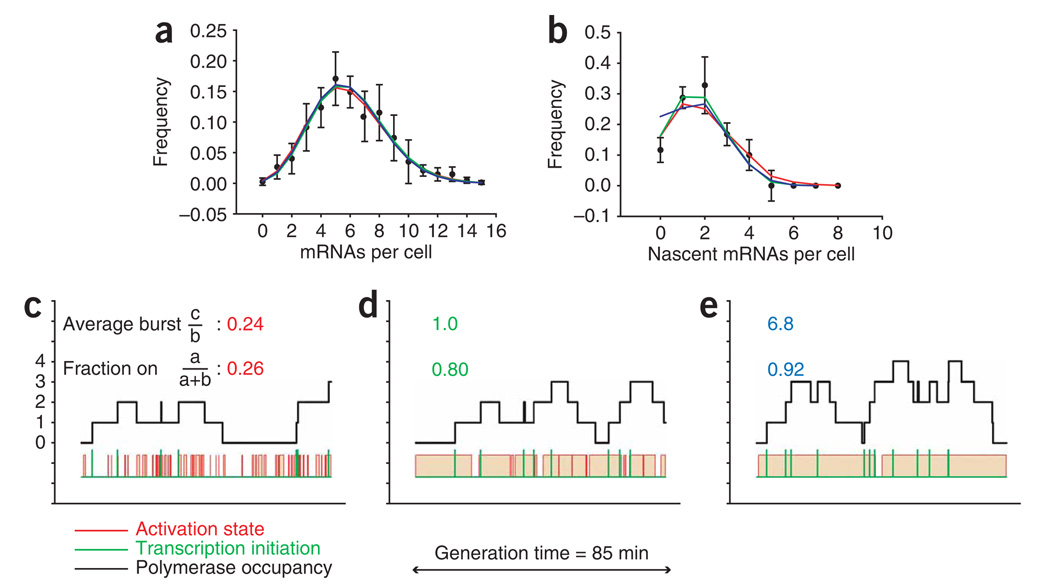
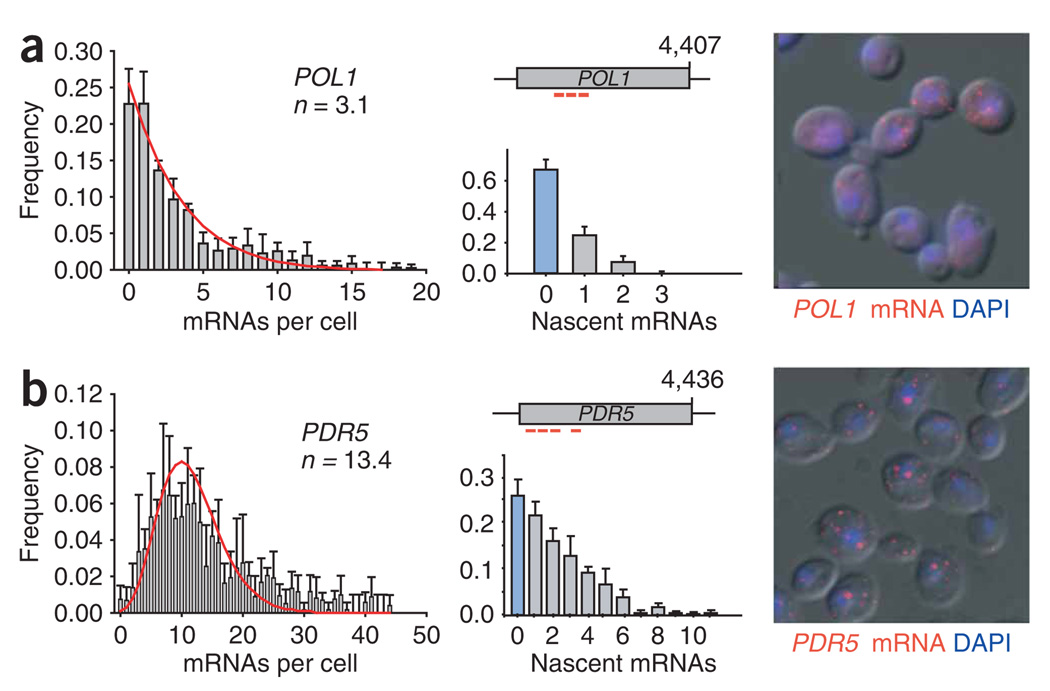
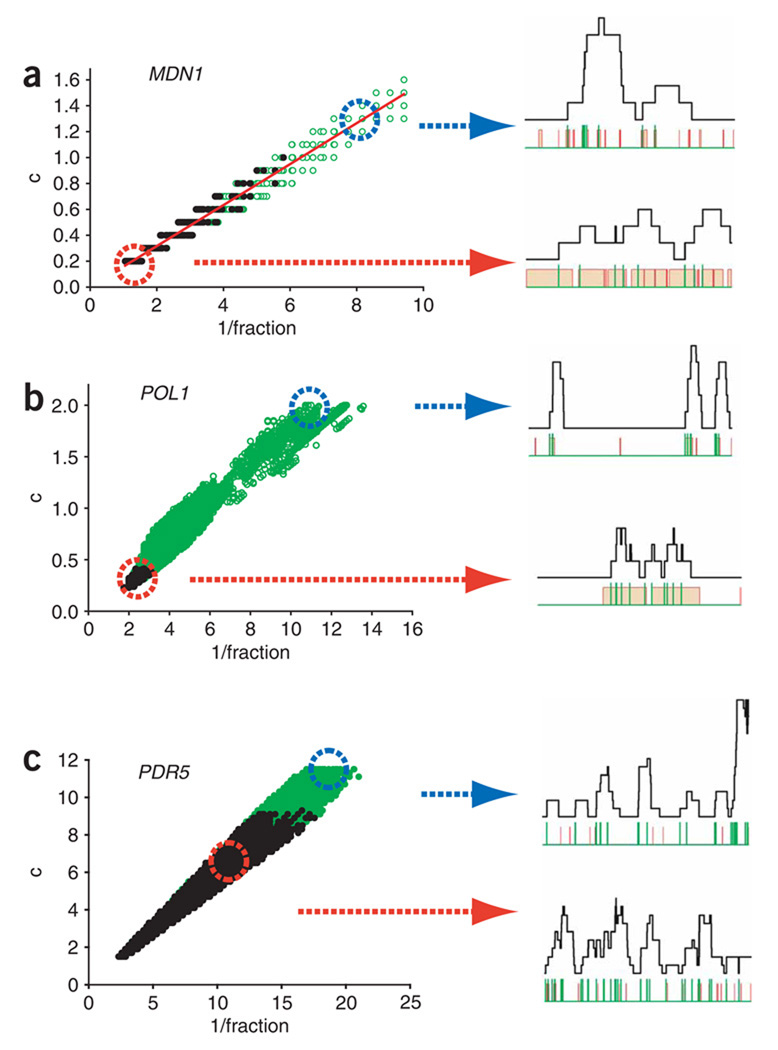
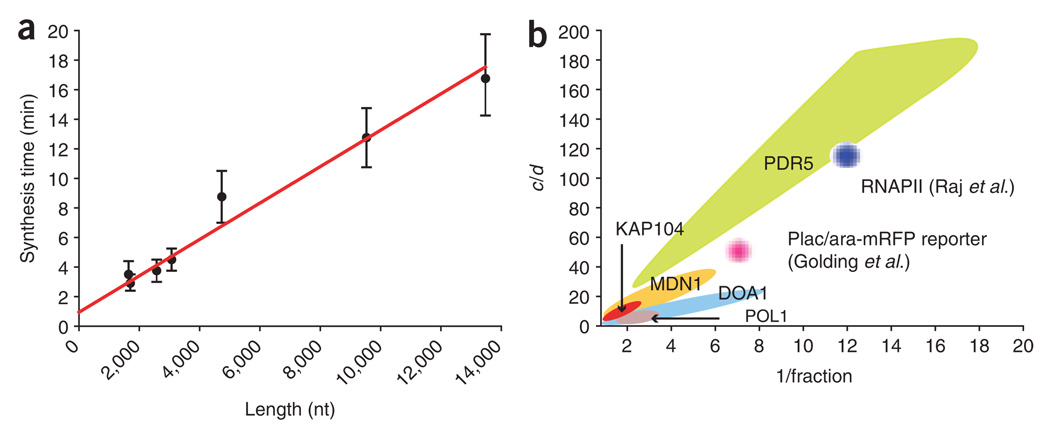
Proper execution of transcriptional programs is a key requirement of gene expression regulation, demanding accurate control of timing and amplitude. How precisely the transcription machinery fulfills this task is not known. Using an in situ hybridization approach that detects single mRNA molecules, we measured mRNA abundance and transcriptional activity within single Saccharomyces cerevisiae cells. We found that expression levels for particular genes are higher than initially reported and can vary substantially among cells. However, variability for most constitutively expressed genes is unexpectedly small. Combining single-transcript measurements with computational modeling indicates that low expression variation is achieved by transcribing genes using single transcription-initiation events that are clearly separated in time, rather than by transcriptional bursts. In contrast, PDR5, a gene regulated by the transcription coactivator complex SAGA, is expressed using transcription bursts, resulting in larger variation. These data directly demonstrate the existence of multiple expression modes used to modulate the transcriptome.
Figures
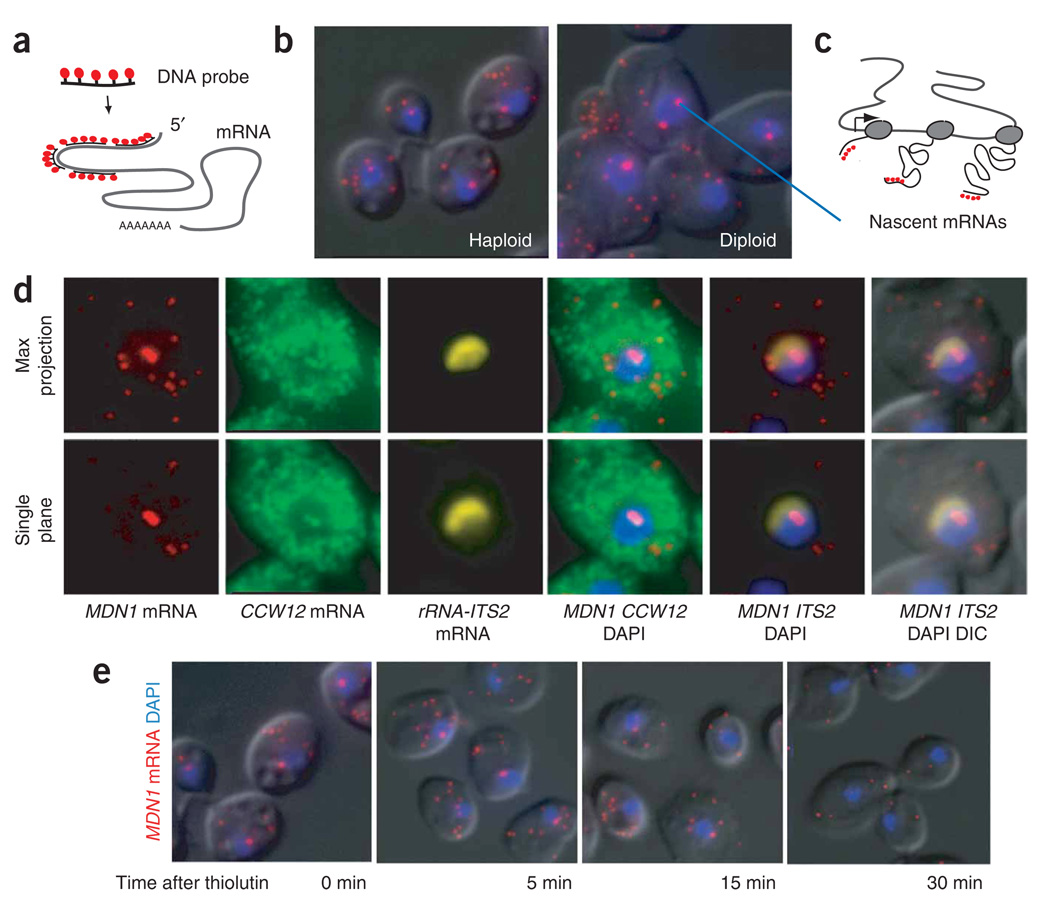
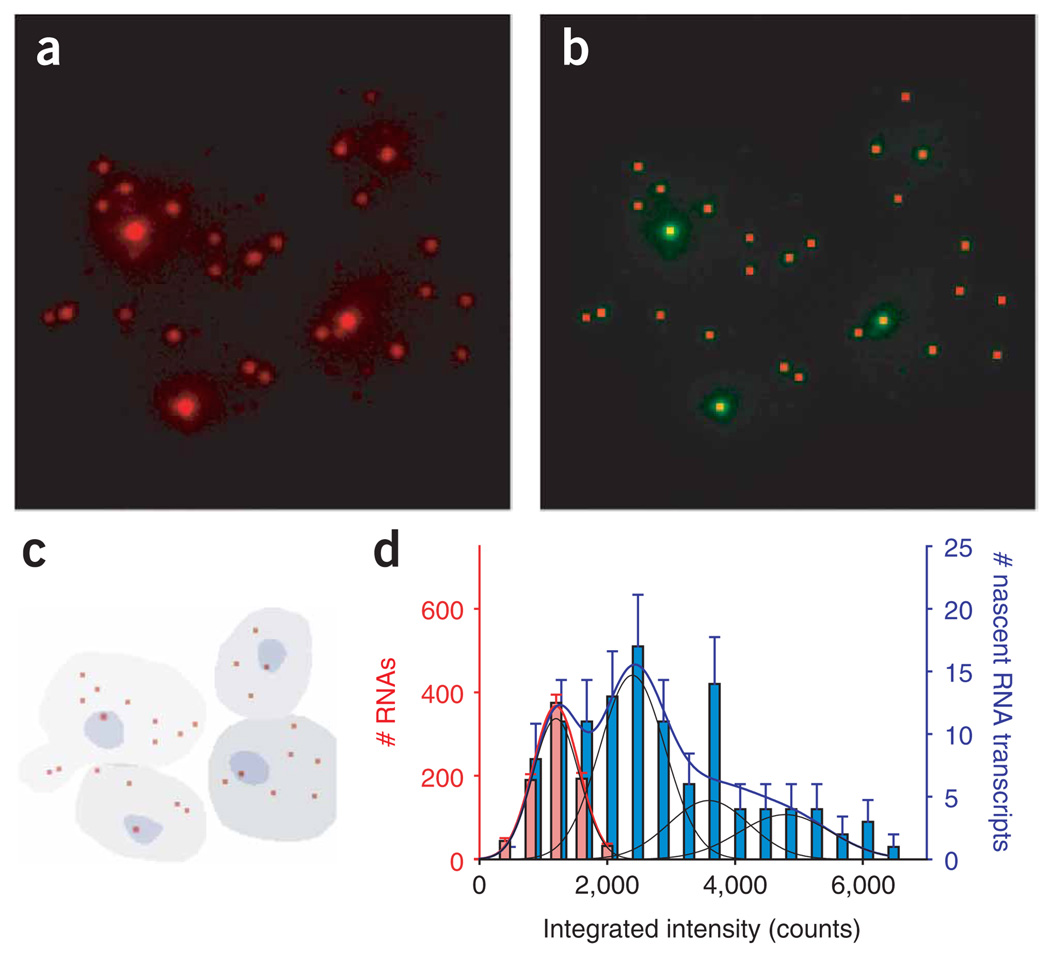
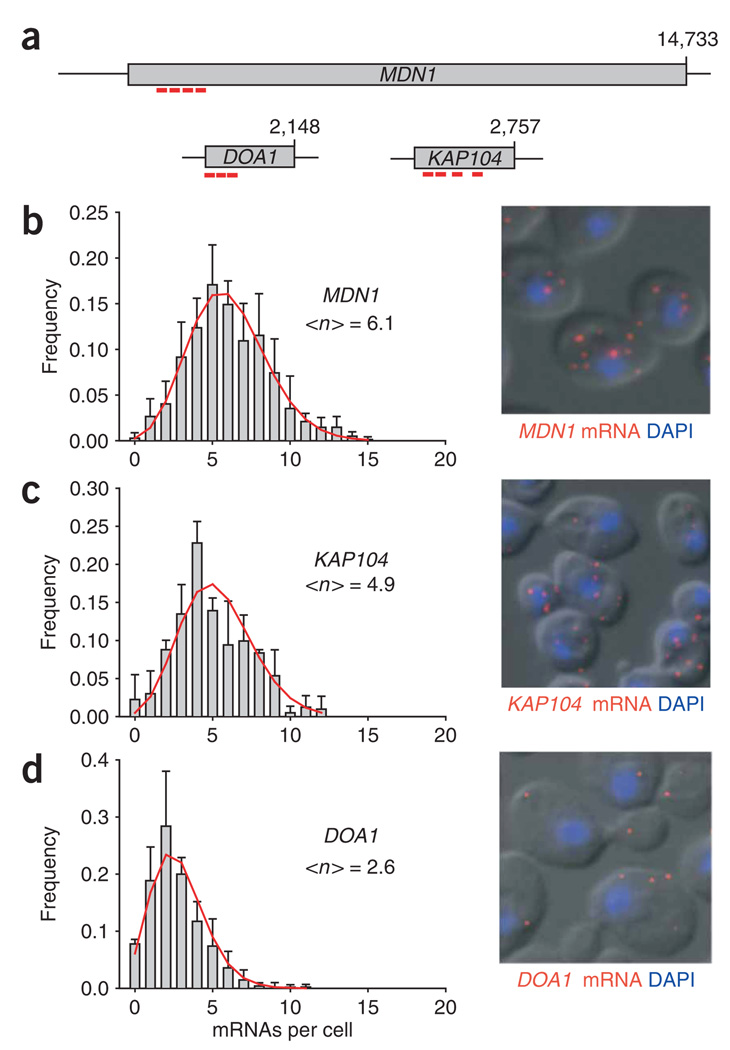
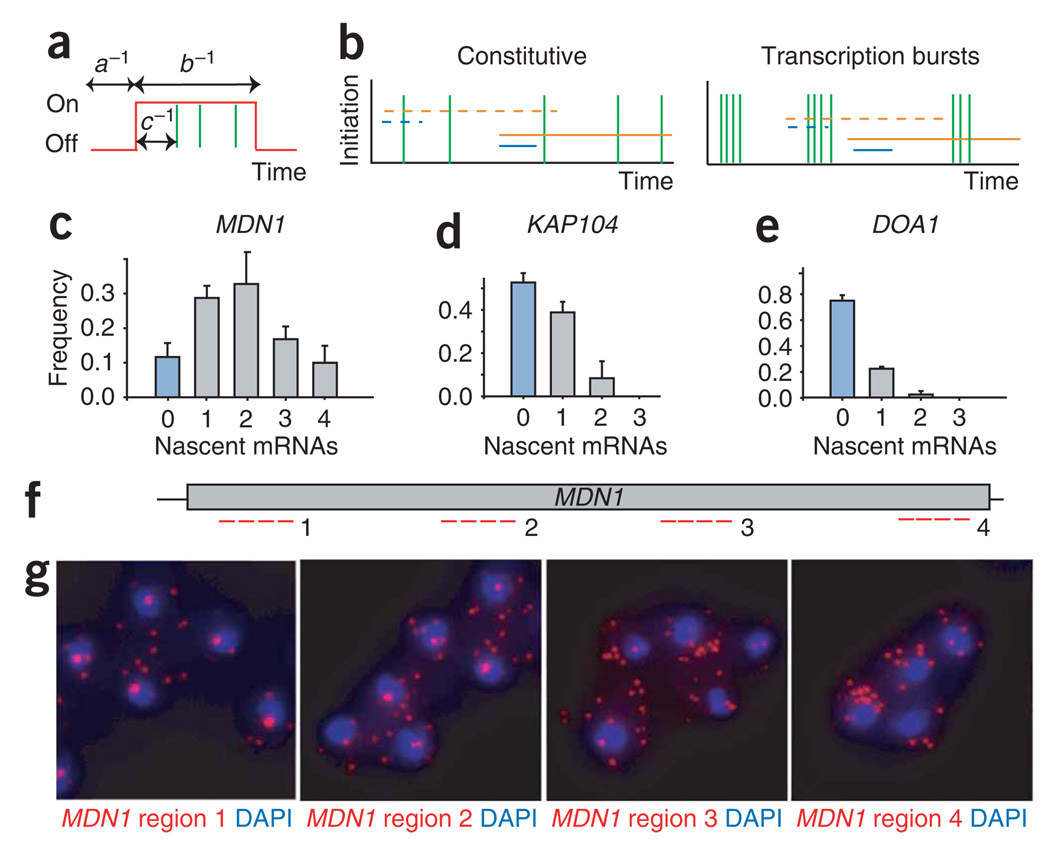
References
-
- Orphanides G, Reinberg D. A unified theory of gene expression. Cell. 2002;108:439–451. - PubMed
-
- Thomas MC, Chiang C-M. Thegeneral transcription machinery and general cofactors. Crit. Rev. Biochem. Mol. Biol. 2006;41:105–178. - PubMed
-
- Dieci G, Sentenac A. Detours and shortcuts to transcription reinitiation. Trends Biochem. Sci. 2003;28:202–209. - PubMed
-
- Li B, Carey M, Workman JL. The role of chromatin during transcription. Cell. 2007;128:707–719. - PubMed
-
- Saunders A, Core LJ, Lis JT. Breaking barriers to transcription elongation. Nat. Rev. Mol. Cell Biol. 2006;7:557–567. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
