

RNAalifold: improved consensus structure prediction for RNA alignments
- PMID: 19014431
- PMCID: PMC2621365
- DOI: 10.1186/1471-2105-9-474
RNAalifold: improved consensus structure prediction for RNA alignments
Abstract
Background: The prediction of a consensus structure for a set of related RNAs is an important first step for subsequent analyses. RNAalifold, which computes the minimum energy structure that is simultaneously formed by a set of aligned sequences, is one of the oldest and most widely used tools for this task. In recent years, several alternative approaches have been advocated, pointing to several shortcomings of the original RNAalifold approach.
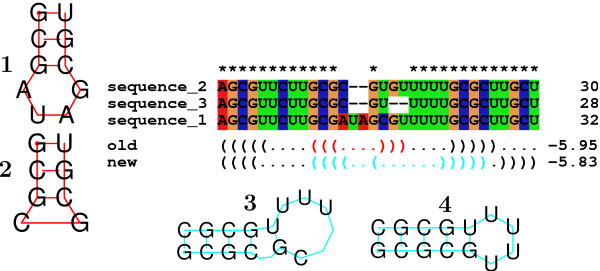
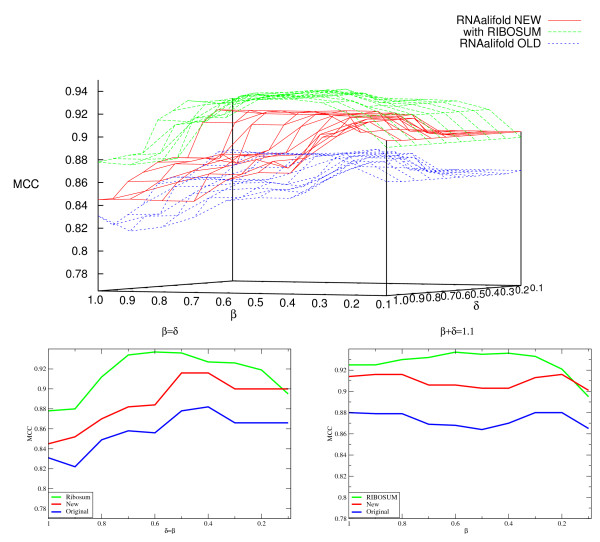
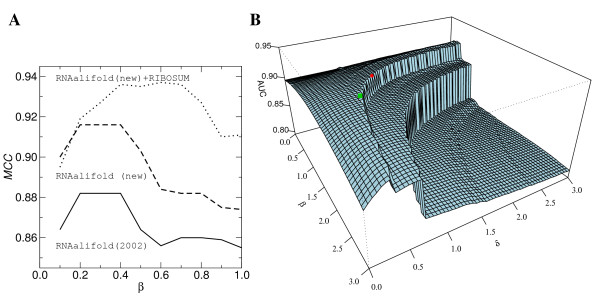
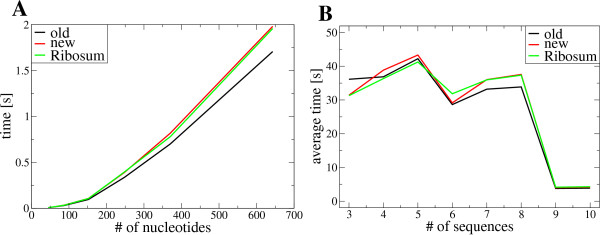
Results: We show that the accuracy of RNAalifold predictions can be improved substantially by introducing a different, more rational handling of alignment gaps, and by replacing the rather simplistic model of covariance scoring with more sophisticated RIBOSUM-like scoring matrices. These improvements are achieved without compromising the computational efficiency of the algorithm. We show here that the new version of RNAalifold not only outperforms the old one, but also several other tools recently developed, on different datasets.
Conclusion: The new version of RNAalifold not only can replace the old one for almost any application but it is also competitive with other approaches including those based on SCFGs, maximum expected accuracy, or hierarchical nearest neighbor classifiers.
Figures
References
-
- Sankoff D. Simultaneous solution of the RNA folding, alignment, and proto-sequence problems. SIAM J Appl Math. 1985;45:810–825. doi: 10.1137/0145048. - DOI
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
