

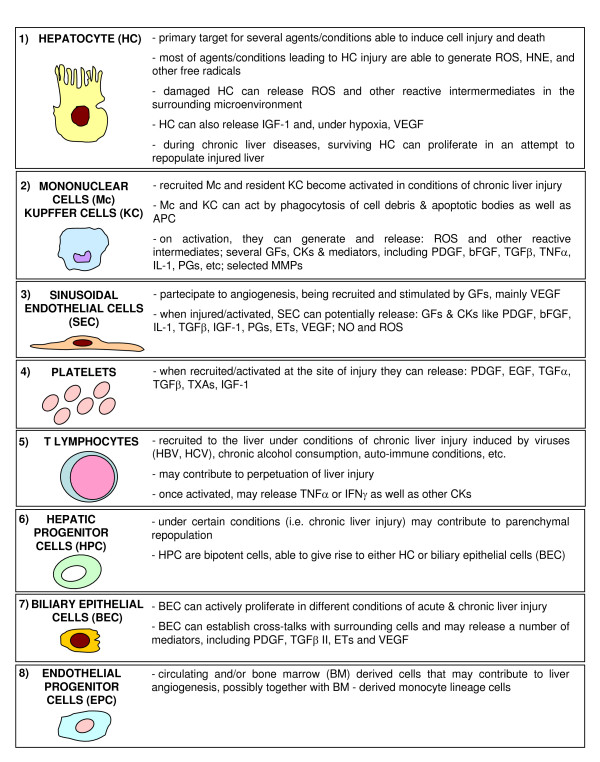
Redox mechanisms in hepatic chronic wound healing and fibrogenesis
- PMID: 19014652
- PMCID: PMC2584013
- DOI: 10.1186/1755-1536-1-5
Redox mechanisms in hepatic chronic wound healing and fibrogenesis
Abstract
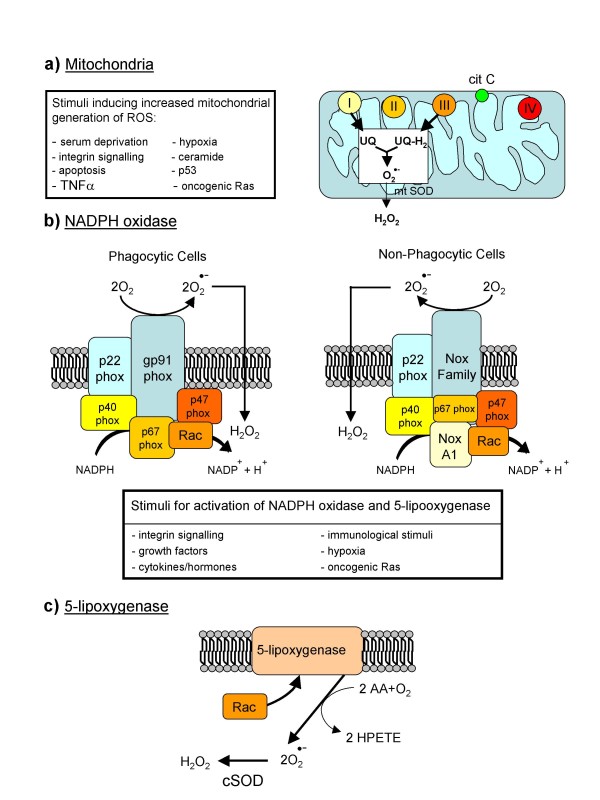
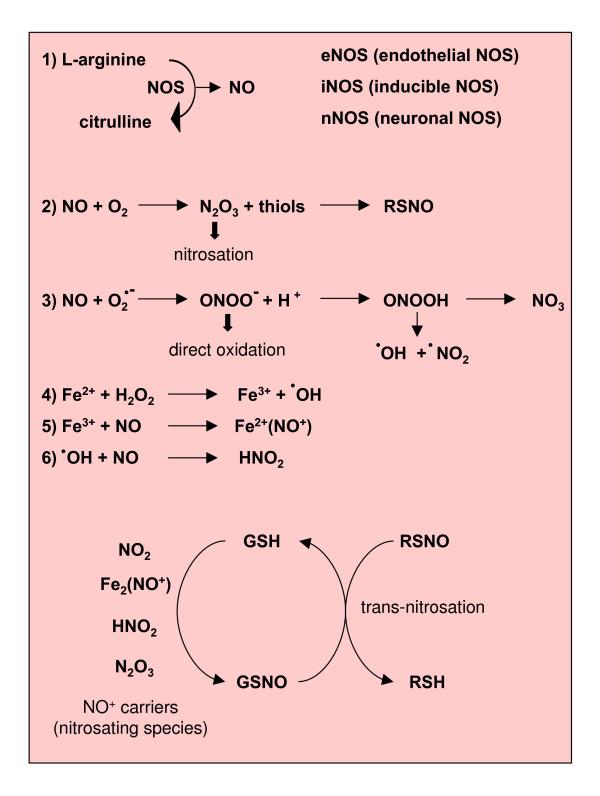
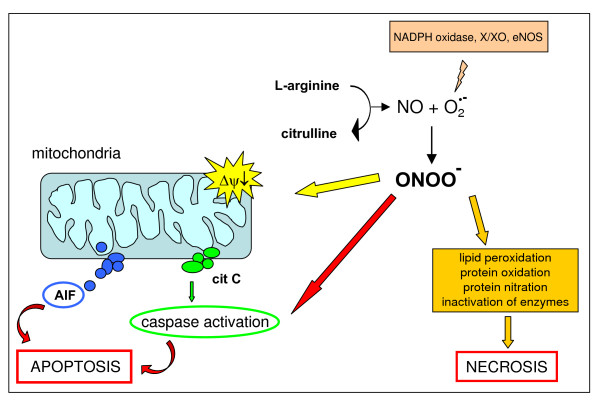
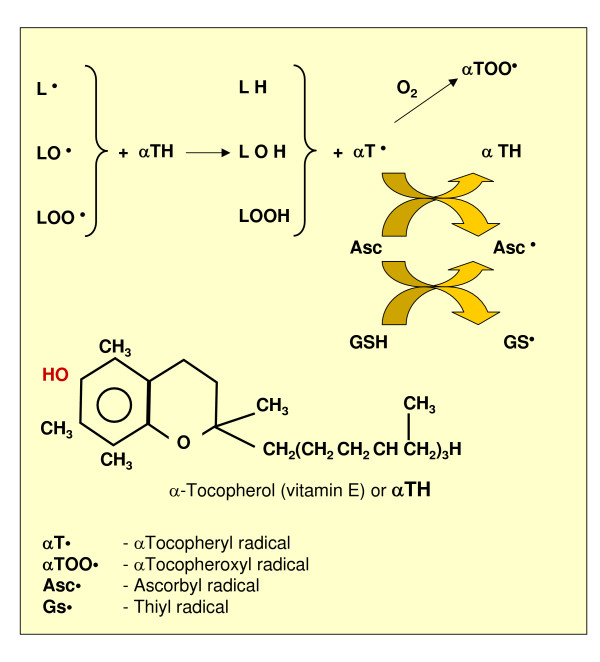
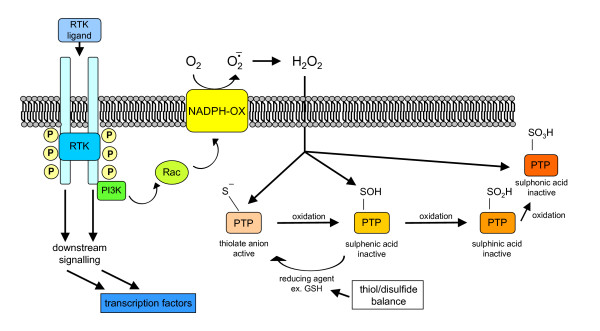
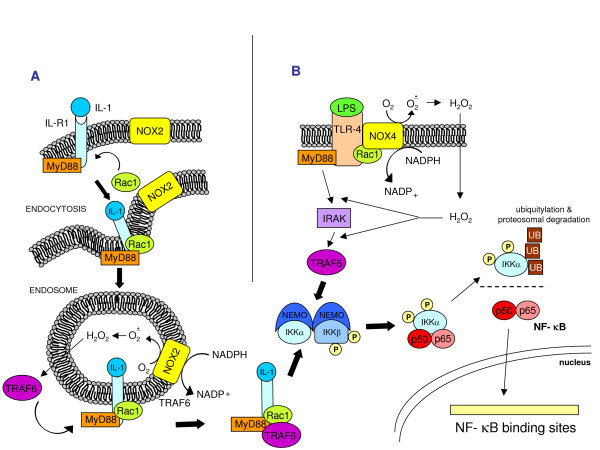
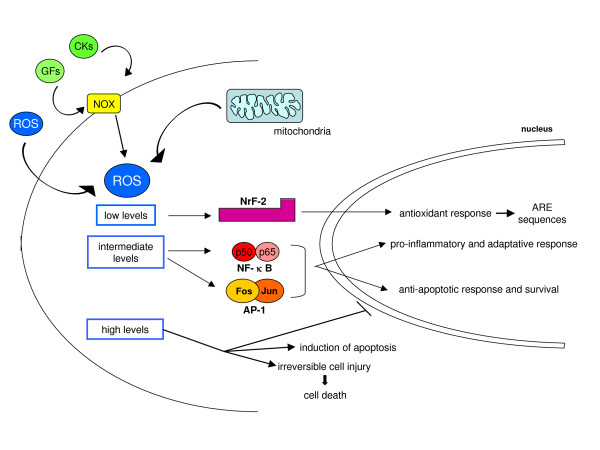
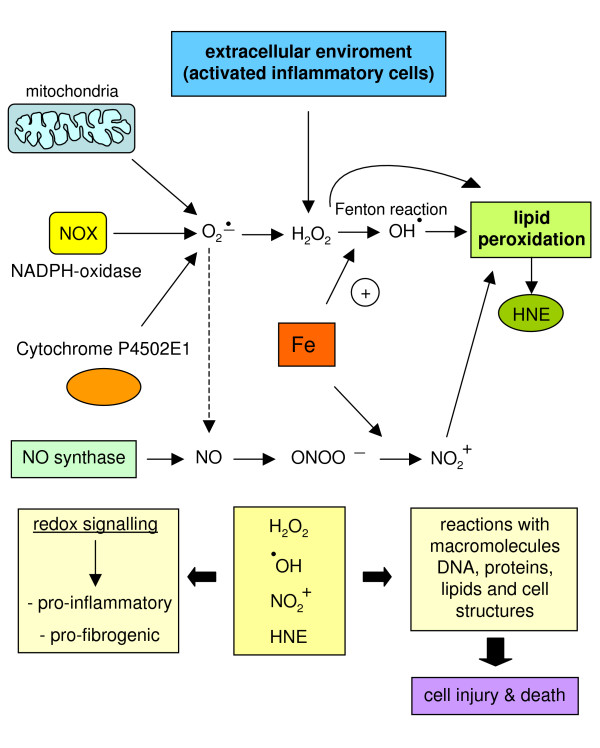
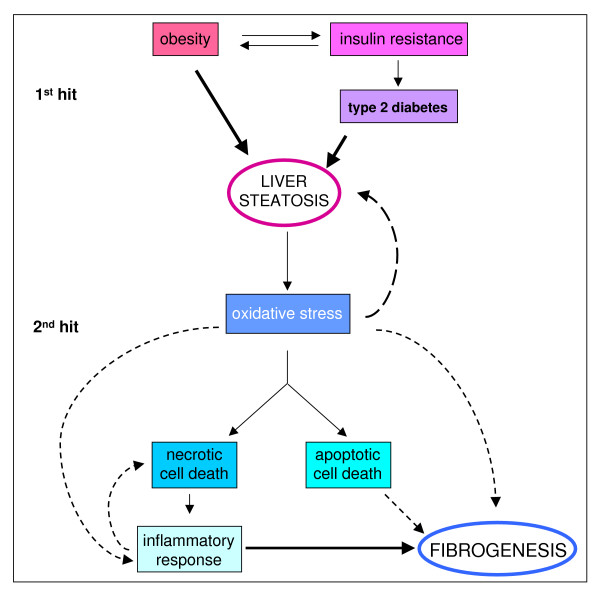
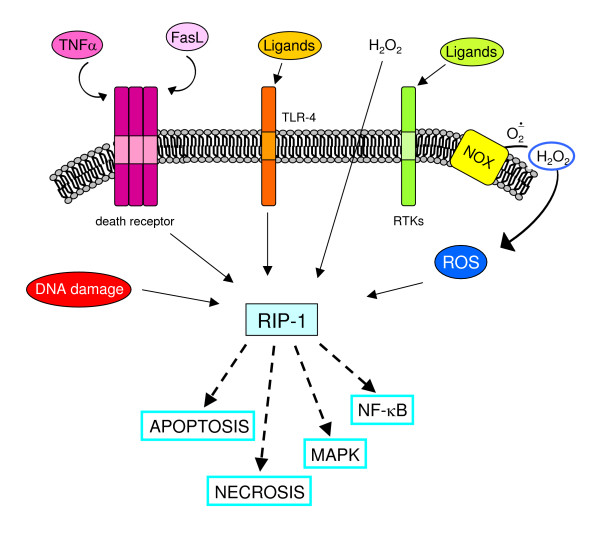
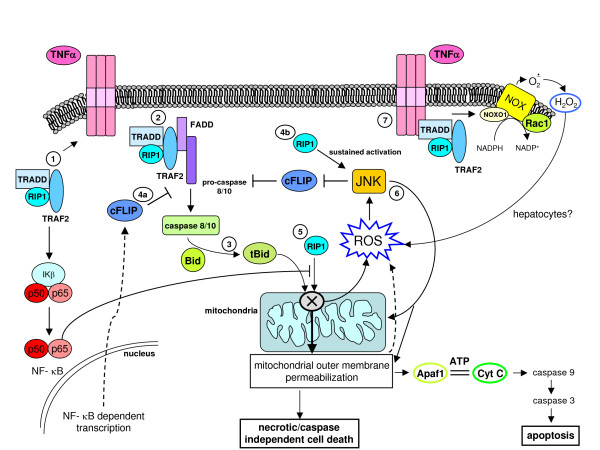
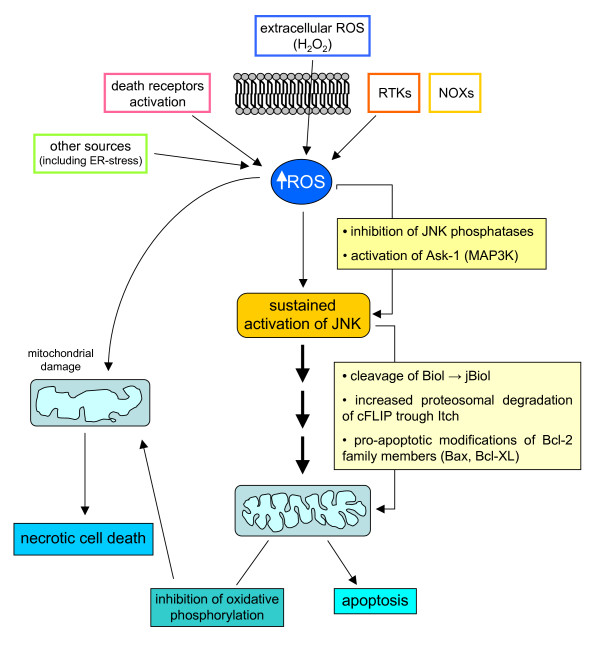
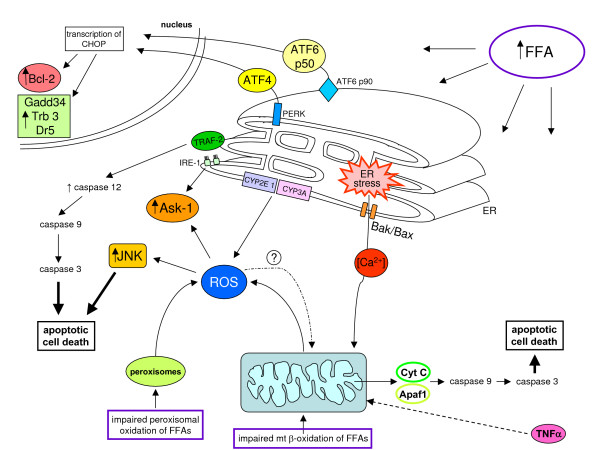
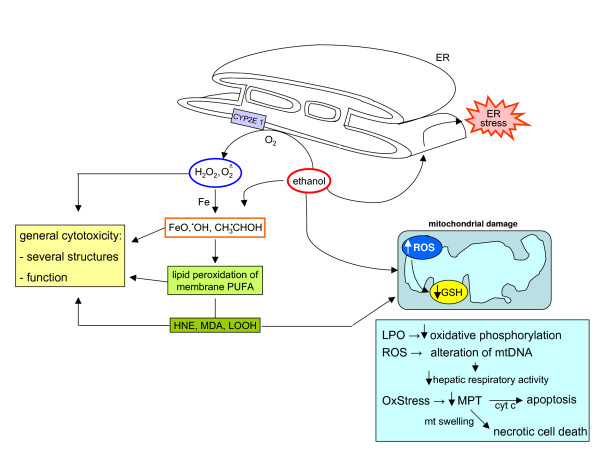
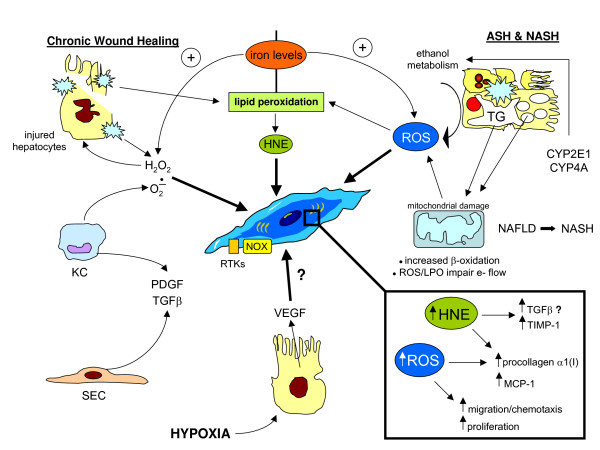
Reactive oxygen species (ROS) generated within cells or, more generally, in a tissue environment, may easily turn into a source of cell and tissue injury. Aerobic organisms have developed evolutionarily conserved mechanisms and strategies to carefully control the generation of ROS and other oxidative stress-related radical or non-radical reactive intermediates (that is, to maintain redox homeostasis), as well as to 'make use' of these molecules under physiological conditions as tools to modulate signal transduction, gene expression and cellular functional responses (that is, redox signalling). However, a derangement in redox homeostasis, resulting in sustained levels of oxidative stress and related mediators, can play a significant role in the pathogenesis of major human diseases characterized by chronic inflammation, chronic activation of wound healing and tissue fibrogenesis. This review has been designed to first offer a critical introduction to current knowledge in the field of redox research in order to introduce readers to the complexity of redox signalling and redox homeostasis. This will include ready-to-use key information and concepts on ROS, free radicals and oxidative stress-related reactive intermediates and reactions, sources of ROS in mammalian cells and tissues, antioxidant defences, redox sensors and, more generally, the major principles of redox signalling and redox-dependent transcriptional regulation of mammalian cells. This information will serve as a basis of knowledge to introduce the role of ROS and other oxidative stress-related intermediates in contributing to essential events, such as the induction of cell death, the perpetuation of chronic inflammatory responses, fibrogenesis and much more, with a major focus on hepatic chronic wound healing and liver fibrogenesis.
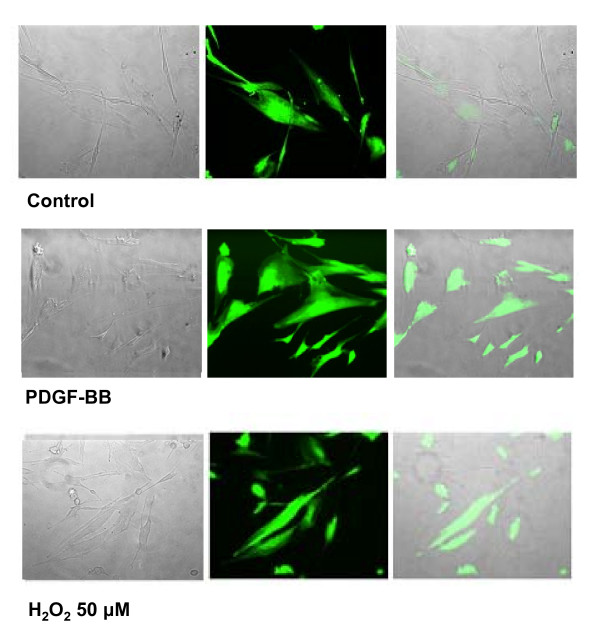
Figures



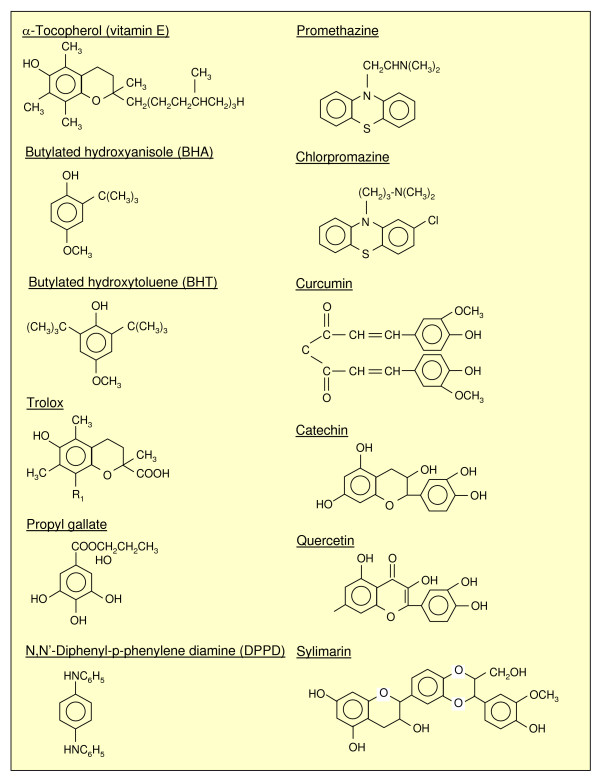
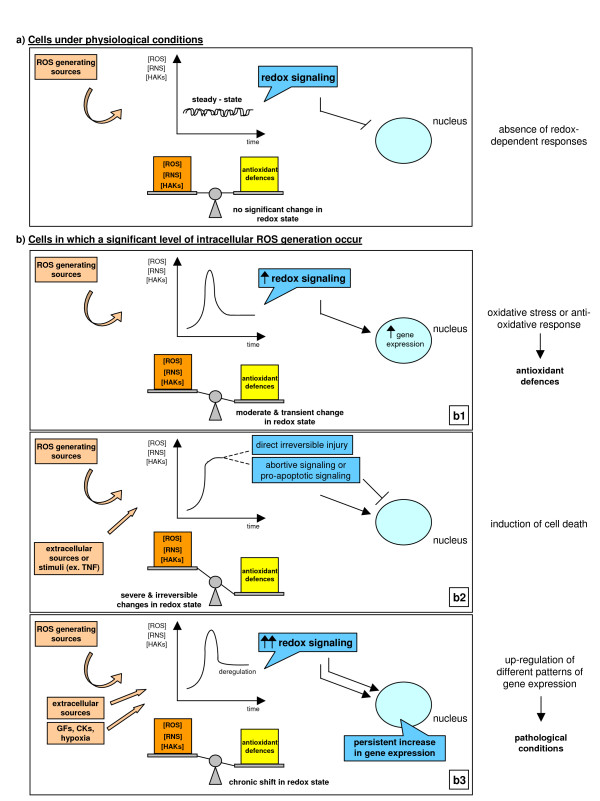
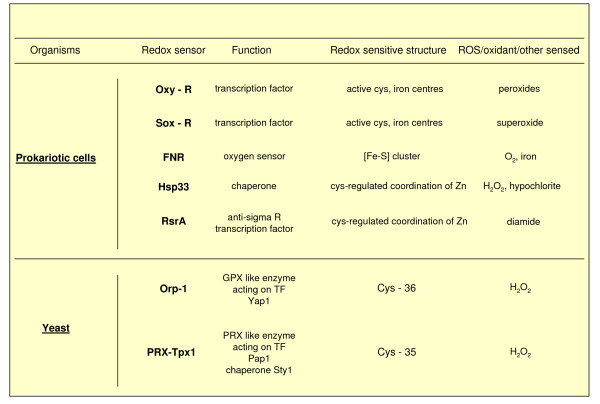
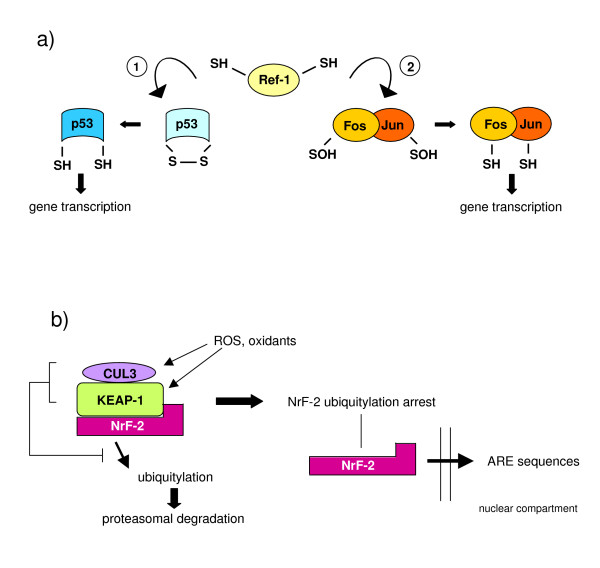
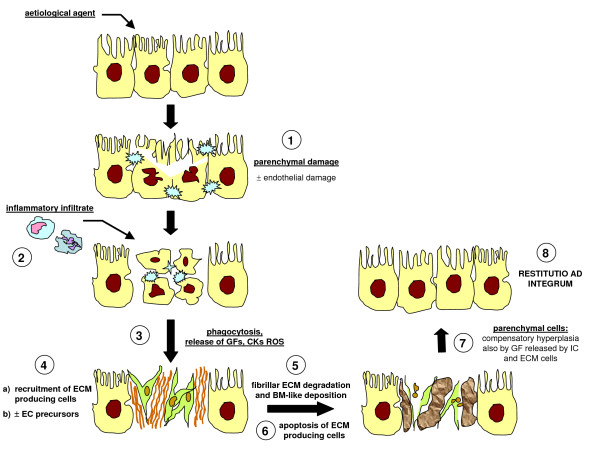
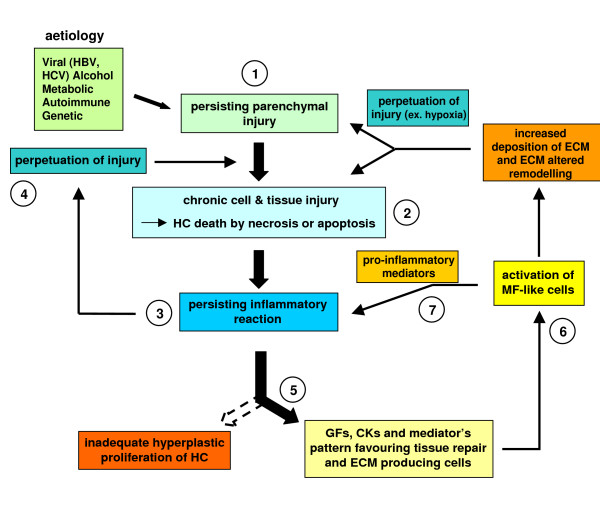
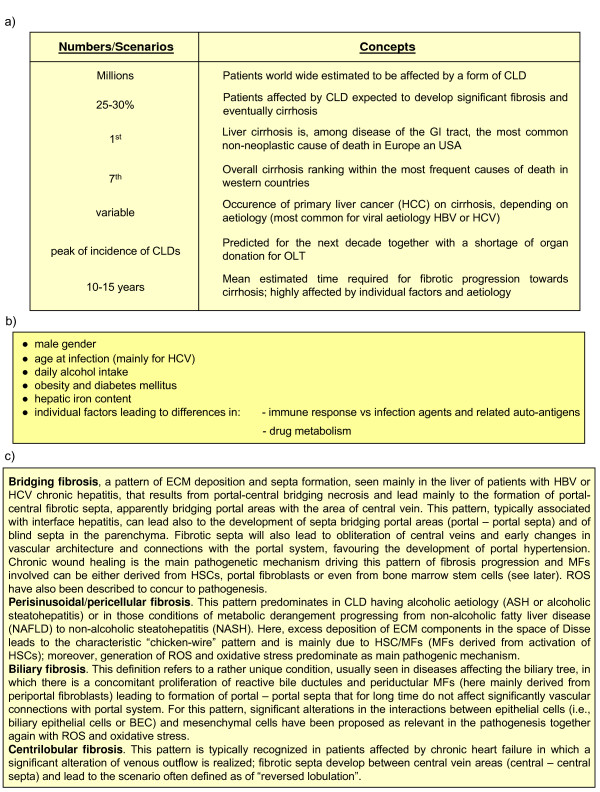
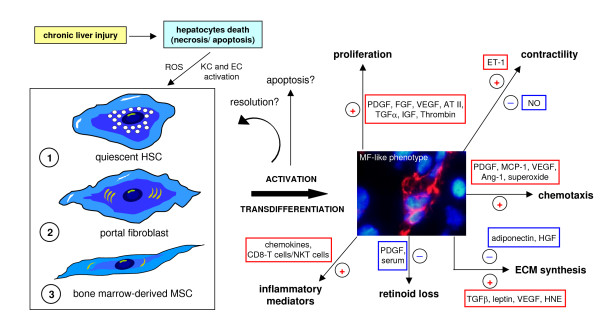

References
LinkOut - more resources
Full Text Sources
Other Literature Sources
