

Use of machine learning algorithms to classify binary protein sequences as highly-designable or poorly-designable
- PMID: 19014713
- PMCID: PMC2655094
- DOI: 10.1186/1471-2105-9-487
Use of machine learning algorithms to classify binary protein sequences as highly-designable or poorly-designable
Abstract
Background: By using a standard Support Vector Machine (SVM) with a Sequential Minimal Optimization (SMO) method of training, Naïve Bayes and other machine learning algorithms we are able to distinguish between two classes of protein sequences: those folding to highly-designable conformations, or those folding to poorly- or non-designable conformations.
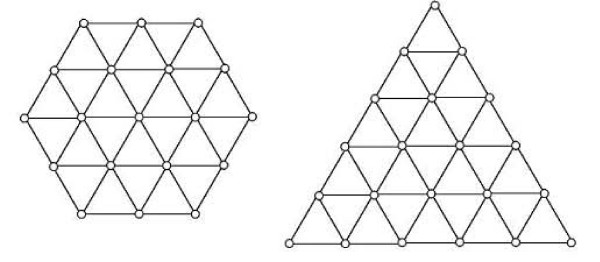
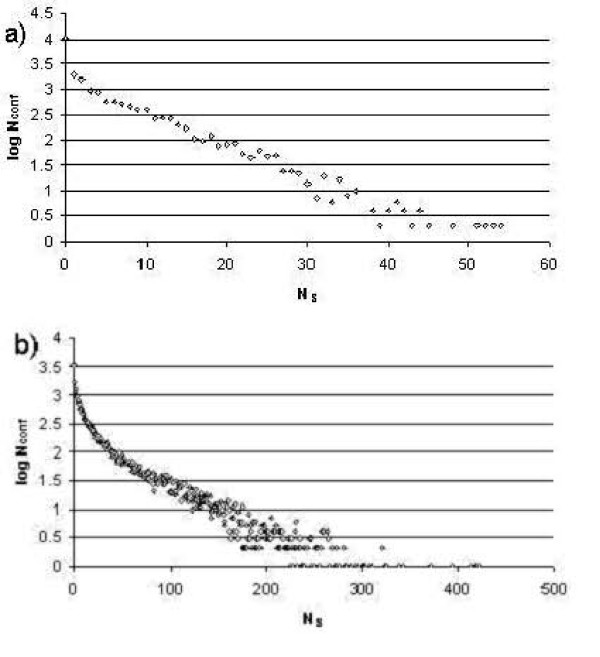
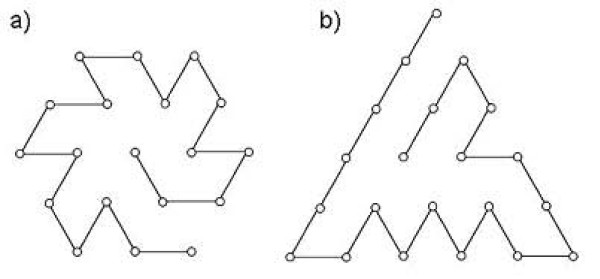
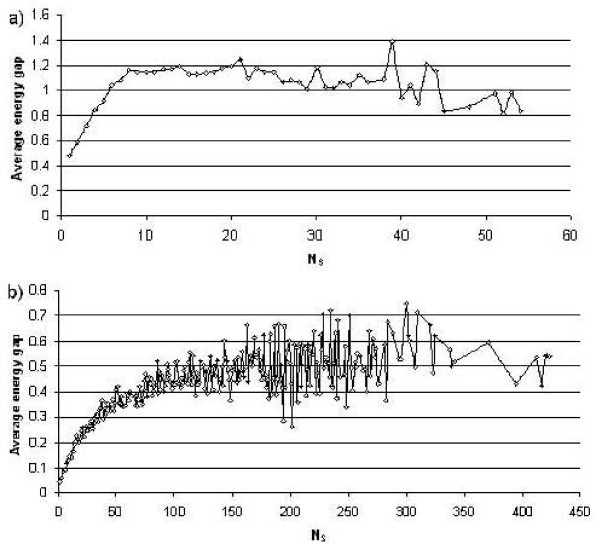
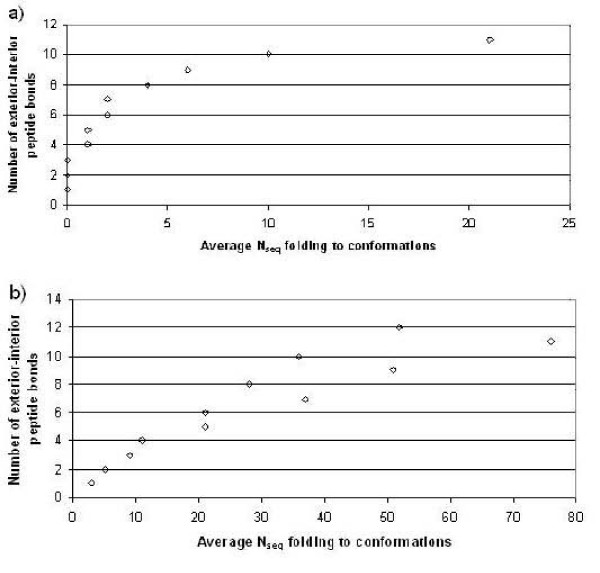
Results: First, we generate all possible compact lattice conformations for the specified shape (a hexagon or a triangle) on the 2D triangular lattice. Then we generate all possible binary hydrophobic/polar (H/P) sequences and by using a specified energy function, thread them through all of these compact conformations. If for a given sequence the lowest energy is obtained for a particular lattice conformation we assume that this sequence folds to that conformation. Highly-designable conformations have many H/P sequences folding to them, while poorly-designable conformations have few or no H/P sequences. We classify sequences as folding to either highly- or poorly-designable conformations. We have randomly selected subsets of the sequences belonging to highly-designable and poorly-designable conformations and used them to train several different standard machine learning algorithms.
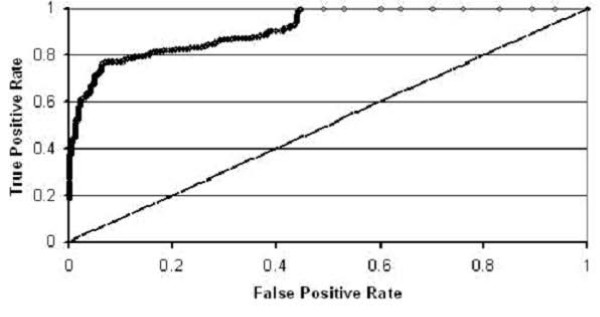
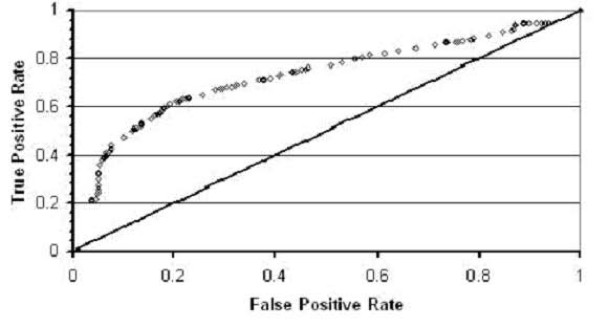
Conclusion: By using these machine learning algorithms with ten-fold cross-validation we are able to classify the two classes of sequences with high accuracy -- in some cases exceeding 95%.
Figures
Similar articles
-
Exploration of the relationship between topology and designability of conformations.J Chem Phys. 2011 Jun 21;134(23):235101. doi: 10.1063/1.3596947. J Chem Phys. 2011. PMID: 21702580 Free PMC article.
-
Predicting Designability of Small Proteins from Graph Features of Contact Maps.J Comput Biol. 2016 May;23(5):400-11. doi: 10.1089/cmb.2015.0209. J Comput Biol. 2016. PMID: 27159634 Free PMC article.
-
Effect of training datasets on support vector machine prediction of protein-protein interactions.Proteomics. 2005 Mar;5(4):876-84. doi: 10.1002/pmic.200401118. Proteomics. 2005. PMID: 15717327
-
Proteins with alternative folds reveal blind spots in AlphaFold-based protein structure prediction.Curr Opin Struct Biol. 2025 Feb;90:102973. doi: 10.1016/j.sbi.2024.102973. Epub 2025 Jan 4. Curr Opin Struct Biol. 2025. PMID: 39756261 Review.
-
Prediction of protein structural class based on symmetrical recurrence quantification analysis.Comput Biol Chem. 2021 Jun;92:107450. doi: 10.1016/j.compbiolchem.2021.107450. Epub 2021 Feb 8. Comput Biol Chem. 2021. PMID: 33631460 Review.
Cited by
-
Exploration of the relationship between topology and designability of conformations.J Chem Phys. 2011 Jun 21;134(23):235101. doi: 10.1063/1.3596947. J Chem Phys. 2011. PMID: 21702580 Free PMC article.
References
-
- Chan HS, Dill KA. The effects of internal constraints on the configurations of chain molecules. J Chem Phys. 1990;92:3118–3135. doi: 10.1063/1.458605. - DOI
-
- Chan HS, Dill KA. Compact polymers. Macromolecules. 2003;22:4559. doi: 10.1021/ma00202a031. - DOI
-
- Crippen GM. Enumeration of cubic lattice walks by contact class. J Chem Phys. 2000;112:11065–11068. doi: 10.1063/1.481746. - DOI
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
