

microRNA-133a regulates cardiomyocyte proliferation and suppresses smooth muscle gene expression in the heart
- PMID: 19015276
- PMCID: PMC2600761
- DOI: 10.1101/gad.1738708
microRNA-133a regulates cardiomyocyte proliferation and suppresses smooth muscle gene expression in the heart
Abstract
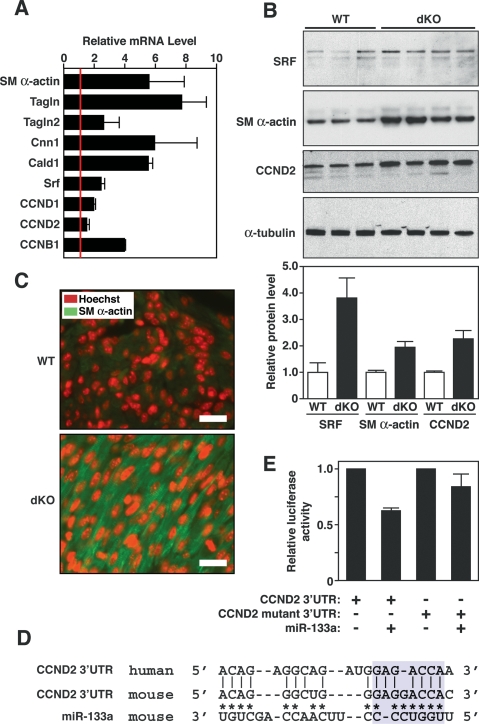
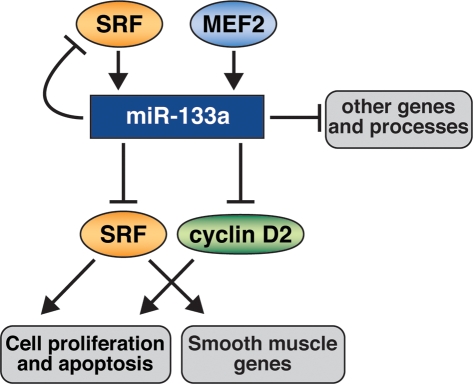
MicroRNAs (miRNAs) modulate gene expression by inhibiting mRNA translation and promoting mRNA degradation, but little is known of their potential roles in organ formation or function. miR-133a-1 and miR-133a-2 are identical, muscle-specific miRNAs that are regulated during muscle development by the SRF transcription factor. We show that mice lacking either miR-133a-1 or miR-133a-2 are normal, whereas deletion of both miRNAs causes lethal ventricular-septal defects in approximately half of double-mutant embryos or neonates; miR-133a double-mutant mice that survive to adulthood succumb to dilated cardiomyopathy and heart failure. The absence of miR-133a expression results in ectopic expression of smooth muscle genes in the heart and aberrant cardiomyocyte proliferation. These abnormalities can be attributed, at least in part, to elevated expression of SRF and cyclin D2, which are targets for repression by miR-133a. These findings reveal essential and redundant roles for miR-133a-1 and miR-133a-2 in orchestrating cardiac development, gene expression, and function and point to these miRNAs as critical components of an SRF-dependent myogenic transcriptional circuit.
Figures

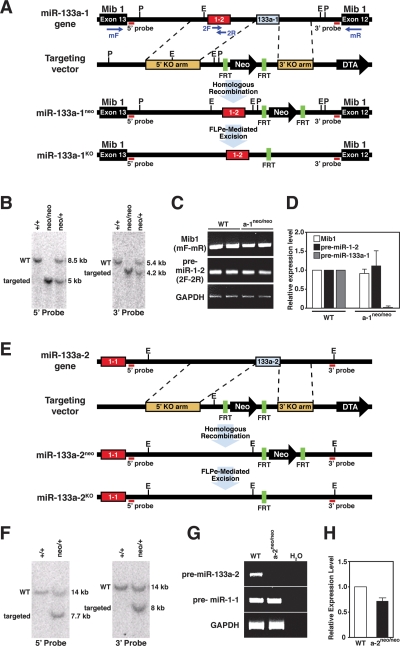
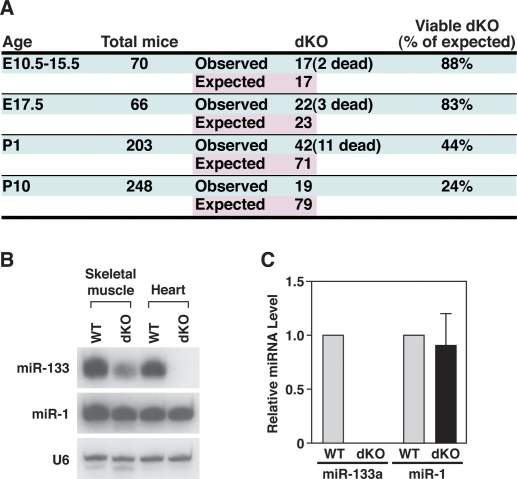
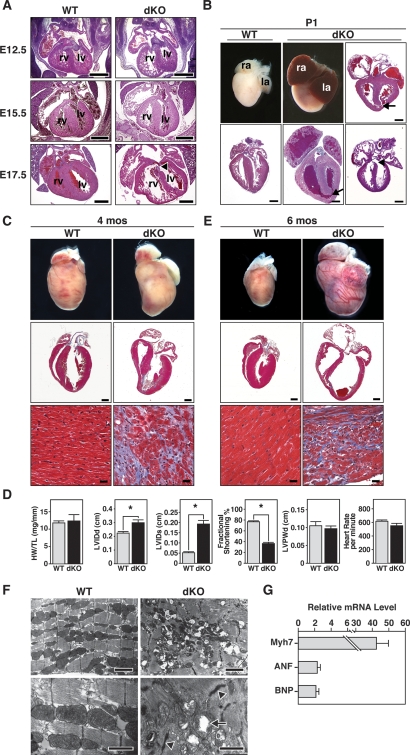
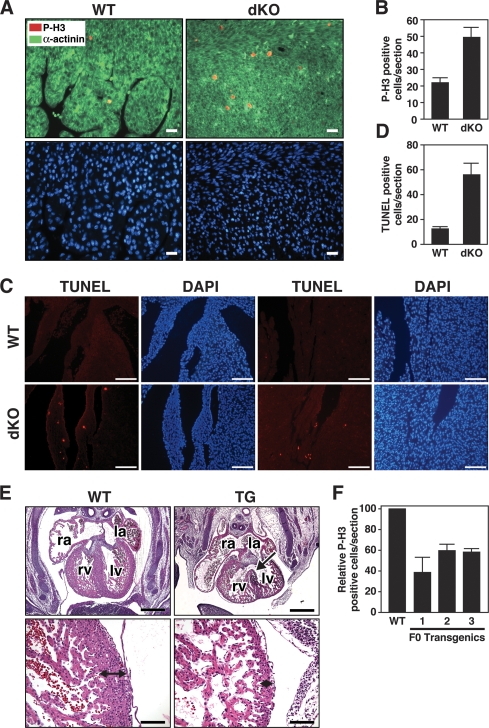
Comment in
-
Right into the heart of microRNA-133a.Genes Dev. 2008 Dec 1;22(23):3227-31. doi: 10.1101/gad.1753508. Genes Dev. 2008. PMID: 19056878 Free PMC article.
References
-
- Care A., Catalucci D., Felicetti F., Bonci D., Addario A., Gallo P., Bang M.L., Segnalini P., Gu Y., Dalton N.D., et al. MicroRNA-133 controls cardiac hypertrophy. Nat. Med. 2007;13:613–618. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous