

SOCS-1 mimetics protect mice against lethal poxvirus infection: identification of a novel endogenous antiviral system
- PMID: 19019946
- PMCID: PMC2620917
- DOI: 10.1128/JVI.01138-08
SOCS-1 mimetics protect mice against lethal poxvirus infection: identification of a novel endogenous antiviral system
Abstract
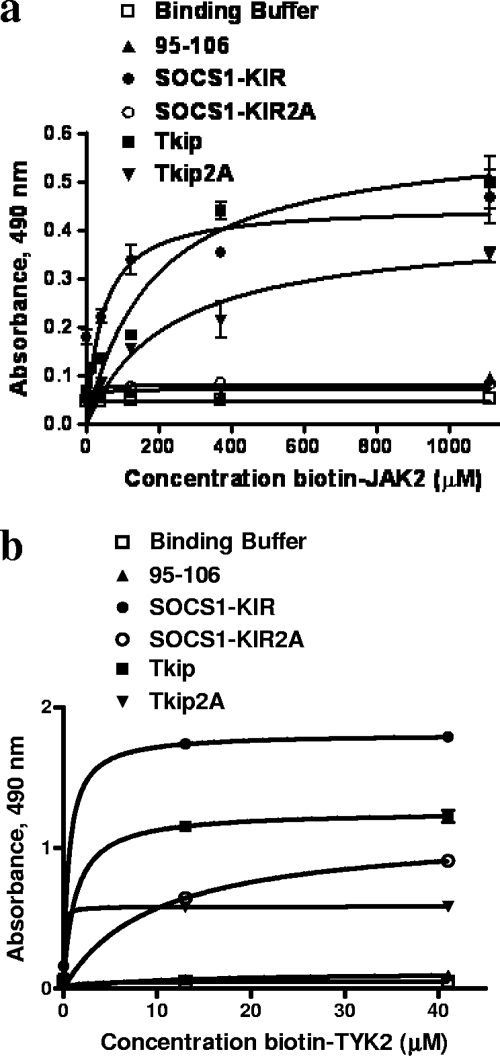
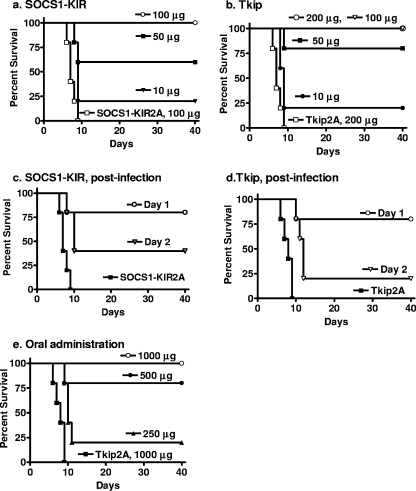
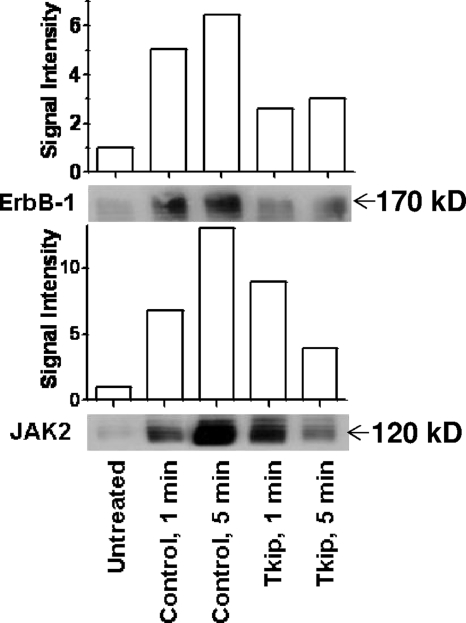
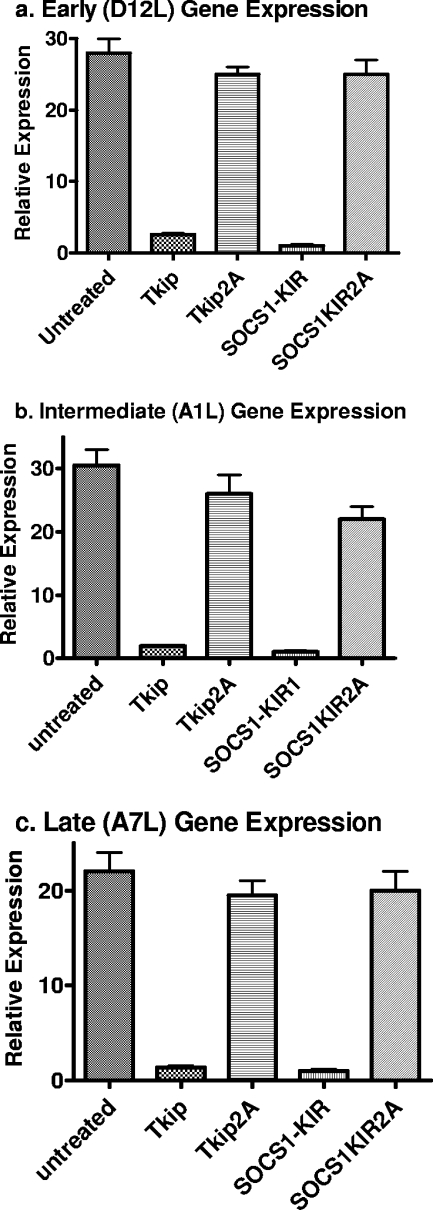
The suppressor of cytokine signaling 1 (SOCS-1) protein modulates cytokine signaling by binding to and inhibiting the function of Janus kinases (JAKs), ErbB, and other tyrosine kinases. We have developed a small tyrosine kinase inhibitor peptide (Tkip) that binds to the autophosphorylation site of tyrosine kinases and inhibits activation of STAT transcription factors. We have also shown that a peptide corresponding to the kinase-inhibitory region of SOCS-1, SOCS1-KIR, similarly interacts with the activation loop of JAK2 and blocks STAT activation. Poxviruses activate cellular tyrosine kinases, such as ErbB-1 and JAK2, in the infection of cells. We used the pathogenesis of vaccinia virus in C57BL/6 mice to determine the ability of the SOCS-1 mimetics to protect mice against lethal vaccinia virus infection. Injection of mice intraperitoneally with Tkip or SOCS1-KIR containing a palmitate for cell penetration, before and at the time of intranasal challenge with 2 x 10(6) PFU of vaccinia virus, resulted in complete protection at 100 microg. Initiation of treatment 1 day postinfection resulted in 80% survival. Administration of SOCS-1 mimetics by the oral route also protected mice against lethal effects of the virus. Both SOCS1-KIR and Tkip inhibited vaccinia virus transcription and replication at early and possibly later stages of infection. Vaccinia virus-induced phosphorylation of ErbB-1 and JAK2 was inhibited by the mimetics. Protected mice mounted a strong humoral and cellular response to vaccinia virus. The use of SOCS-1 mimetics in the treatment of poxvirus infections reveals an endogenous regulatory system that previously was not known to have an antiviral function.
Figures







References
-
- Ahmed, C. M. I., and H. M. Johnson. 2006. IFN-γ and its receptor subunit IFNGR1 are recruited to the IFN-γ-activated sequence element at the promoter site of IFN-γ-activated genes: evidence of transactivational activity in IFNGR1. J. Immunol. 177315-321. - PubMed
-
- Ahmed, C. M., J. P. Martin, and H. M. Johnson. 2007. IFN mimetic as a therapeutic for lethal vaccinia virus infection: possible effects on innate and adaptive immune responses. J. Immunol. 1784576-4583. - PubMed
-
- Alexander, W. S., and D. J. Hilton. 2004. The role of suppressors of cytokine signaling (SOCS) proteins in regulation of immune response. Annu. Rev. Immunol. 22503-529. - PubMed
-
- Ausubel, F. M., R. Brent, R. E. Kingston, D. D. Moore, J. G. Seidman, A. Smith, and K. Struhl (ed.). 2002. Short protocols in molecular biology, p. 16-65. Wiley, New York, NY.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
