

Molecular mechanisms of collagen isotype-specific modulation of smooth muscle cell phenotype
- PMID: 19023090
- PMCID: PMC2692987
- DOI: 10.1161/ATVBAHA.108.178749
Molecular mechanisms of collagen isotype-specific modulation of smooth muscle cell phenotype
Abstract
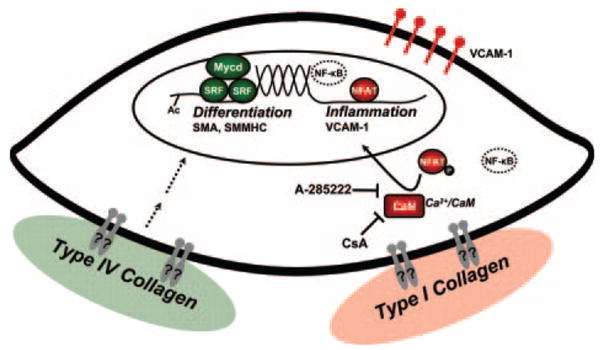
Objective: Smooth muscle cell (SMC) phenotypic modulation, an important component of atherosclerosis progression, is critically regulated by the matrix, with normal components of the healthy SMC matrix limiting modulation and atherosclerosis-associated transitional matrix proteins promoting phenotypic modulation. We sought to determine how collagen IV (which comprises the healthy artery wall) and monomeric collagen I (which comprises atherosclerotic lesions) differentially affect SMC phenotype.
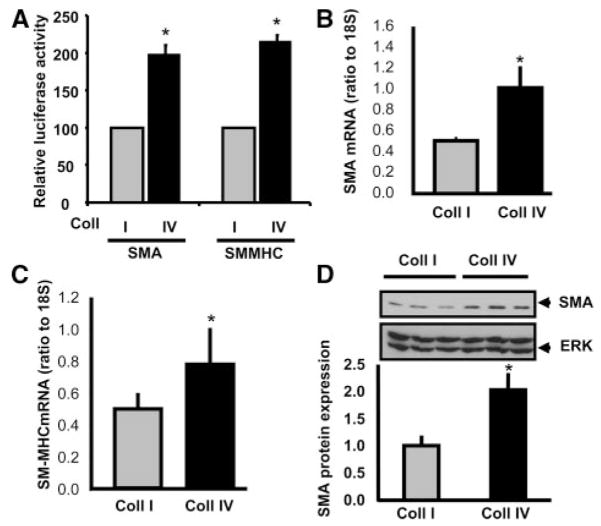
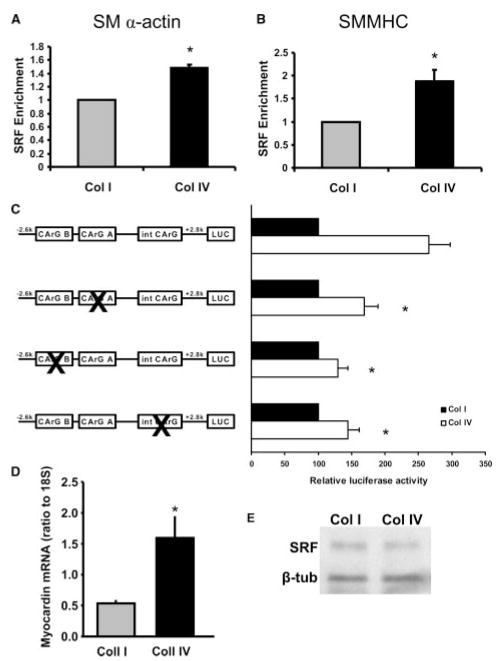
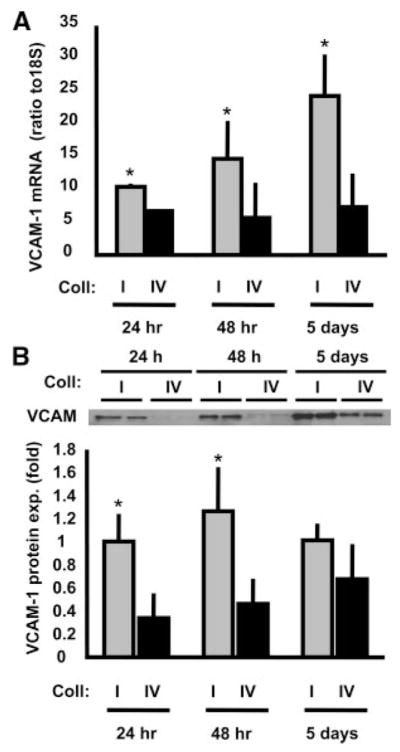
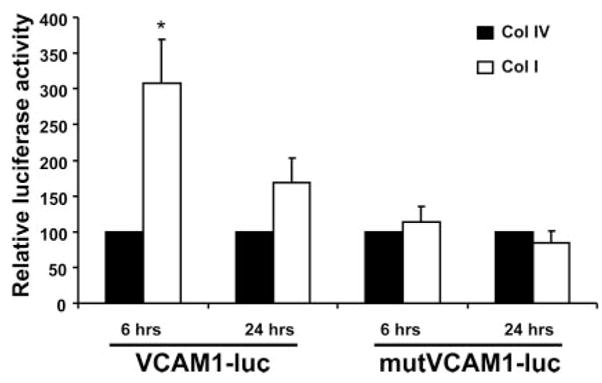
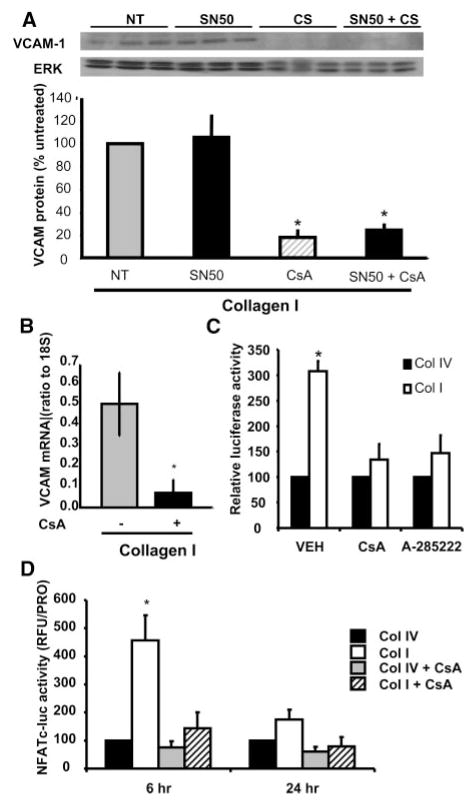
Methods and results: Plating SMCs on collagen IV resulted in elevated expression of SMC contractility proteins compared to collagen I. Concurrent with enhanced contractile gene expression, collagen IV stimulates binding of SRF to CArG boxes in the promoters of smooth muscle actin and smooth muscle myosin heavy chain. Coll IV also stimulated the expression of myocardin, a critical SRF coactivator required to drive expression of SMC specific genes. In contrast to collagen IV, collagen I stimulated enhanced expression of the inflammatory protein vascular cell adhesion molecule (VCAM)-1. NF-kappaB and NFAT-binding sites in the VCAM-1 promoter are critical for collagen I-mediated expression of VCAM-1 promoter activity. However, only inhibitors of NFAT, not NF-kappaB, were able to reduce collagen I-associated VCAM expression, and collagen I but not collagen IV stimulated NFAT transcriptional activity.
Conclusions: These results show for the first time that collagen IV and collagen I differentially affect smooth muscle phenotypic modulation through multiple pathways.
Figures
Similar articles
-
Smooth muscle cell-specific transcription is regulated by nuclear localization of the myocardin-related transcription factors.Am J Physiol Heart Circ Physiol. 2007 Feb;292(2):H1170-80. doi: 10.1152/ajpheart.00864.2006. Epub 2006 Sep 22. Am J Physiol Heart Circ Physiol. 2007. PMID: 16997888
-
Drug-eluting stent specifically designed to target vascular smooth muscle cell phenotypic modulation attenuated restenosis through the YAP pathway.Am J Physiol Heart Circ Physiol. 2019 Sep 1;317(3):H541-H551. doi: 10.1152/ajpheart.00089.2019. Epub 2019 Jul 12. Am J Physiol Heart Circ Physiol. 2019. PMID: 31298560
-
L-type voltage-gated Ca2+ channels modulate expression of smooth muscle differentiation marker genes via a rho kinase/myocardin/SRF-dependent mechanism.Circ Res. 2004 Aug 20;95(4):406-14. doi: 10.1161/01.RES.0000138582.36921.9e. Epub 2004 Jul 15. Circ Res. 2004. PMID: 15256479
-
Myocardin: A novel player in atherosclerosis.Atherosclerosis. 2017 Feb;257:266-278. doi: 10.1016/j.atherosclerosis.2016.12.002. Epub 2016 Dec 1. Atherosclerosis. 2017. PMID: 28012646 Review.
-
Transcriptional regulation of vascular smooth muscle cell proliferation, differentiation and senescence: Novel targets for therapy.Vascul Pharmacol. 2022 Oct;146:107091. doi: 10.1016/j.vph.2022.107091. Epub 2022 Jul 25. Vascul Pharmacol. 2022. PMID: 35896140 Review.
Cited by
-
Differentiation of Murine Bone Marrow-Derived Smooth Muscle Progenitor Cells Is Regulated by PDGF-BB and Collagen.PLoS One. 2016 Jun 3;11(6):e0156935. doi: 10.1371/journal.pone.0156935. eCollection 2016. PLoS One. 2016. PMID: 27258003 Free PMC article.
-
Collagen inhibitory peptide R1R2 mediates vascular remodeling by decreasing inflammation and smooth muscle cell activation.PLoS One. 2015 Feb 12;10(2):e0117356. doi: 10.1371/journal.pone.0117356. eCollection 2015. PLoS One. 2015. PMID: 25675397 Free PMC article.
-
Crosstalk Between Macrophages and Vascular Smooth Muscle Cells in Atherosclerotic Plaque Stability.Arterioscler Thromb Vasc Biol. 2022 Apr;42(4):372-380. doi: 10.1161/ATVBAHA.121.316233. Epub 2022 Feb 17. Arterioscler Thromb Vasc Biol. 2022. PMID: 35172605 Free PMC article. Review.
-
Radial Artery Tonometry is Associated With Major Adverse Cardiac Events in Patients With Peripheral Artery Disease.J Surg Res. 2019 Mar;235:250-257. doi: 10.1016/j.jss.2018.09.088. Epub 2018 Nov 1. J Surg Res. 2019. PMID: 30691803 Free PMC article.
-
Integrin signaling in atherosclerosis.Cell Mol Life Sci. 2017 Jun;74(12):2263-2282. doi: 10.1007/s00018-017-2490-4. Epub 2017 Feb 28. Cell Mol Life Sci. 2017. PMID: 28246700 Free PMC article. Review.
References
-
- Yusuf S, Reddy S, Ounpuu S, Anand S. Global burden of cardiovascular diseases: part I: general considerations, the epidemiologic transition, risk factors, and impact of urbanization. Circulation. 2001;104:2746–2753. - PubMed
-
- Ross R. Atherosclerosis–an inflammatory disease.[see comment] N Engl J Med. 1999;340:115–126. - PubMed
-
- Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev. 2004;84:767–801. - PubMed
-
- Owens GK. Regulation of differentiation of vascular smooth muscle cells. Physiol Rev. 1995;75:487–517. - PubMed
-
- Libby P. Inflammation in atherosclerosis. Nature. 2002;420:868–874. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
