

Lactadherin and clearance of platelet-derived microvesicles
- PMID: 19023116
- PMCID: PMC2637196
- DOI: 10.1182/blood-2008-07-167148
Lactadherin and clearance of platelet-derived microvesicles
Abstract
The transbilayer movement of phosphatidylserine from the inner to the outer leaflet of the membrane bilayer during platelet activation is associated with the release of procoagulant phosphatidylserine-rich small membrane vesicles called platelet-derived microvesicles. We tested the effect of lactadherin, which promotes the phagocytosis of phosphatidylserine-expressing lymphocytes and red blood cells, in the clearance of platelet microvesicles. Platelet-derived microvesicles were labeled with BODIPY-maleimide and incubated with THP-1-derived macrophages. The extent of phagocytosis was quantified by flow cytometry. Lactadherin promoted phagocytosis in a concentration-dependent manner with a half-maximal effect at approximately 5 ng/mL. Lactadherin-deficient mice had increased number of platelet-derived microvesicles in their plasma compared with their wild-type littermates (950 +/- 165 vs 4760 +/- 650; P = .02) and generated 2-fold more thrombin. In addition, splenic macrophages from lactadherin-deficient mice showed decreased capacity to phagocytose platelet-derived microvesicles. In an in vivo model of light/dye-induced endothelial injury/thrombosis in the cremasteric venules, lactadherin-deficient mice had significantly shorter time for occlusion compared with their wild-type littermate controls (5.93 +/- 0.43 minutes vs 9.80 +/- 1.14 minutes;P = .01). These studies show that lactadherin mediates the clearance of phosphatidylserine-expressing platelet-derived microvesicles from the circulation and that a defective clearance can induce a hypercoagulable state.
Figures
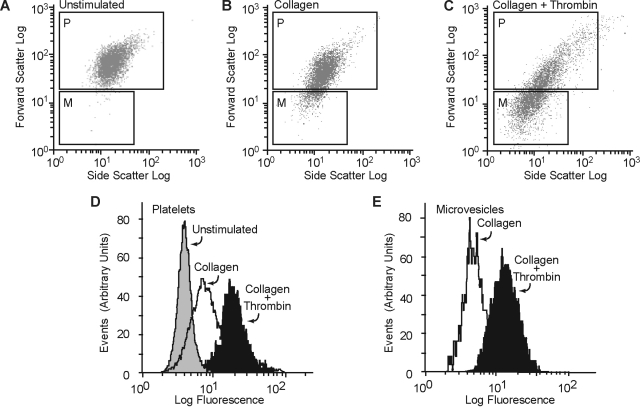
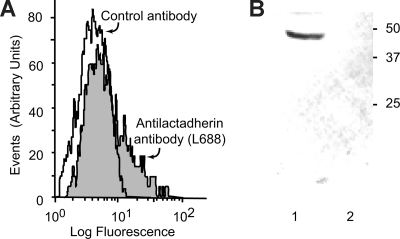
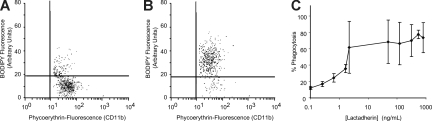
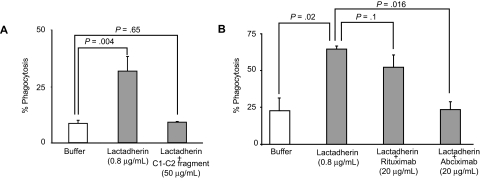
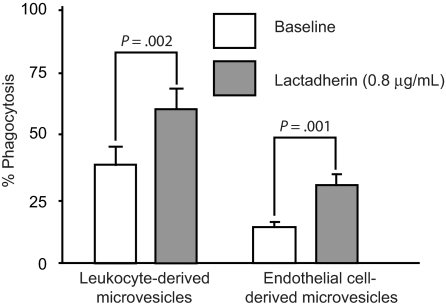
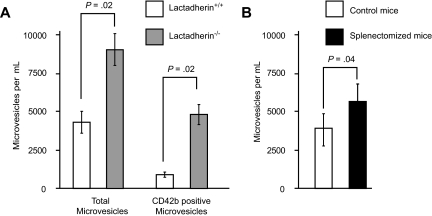
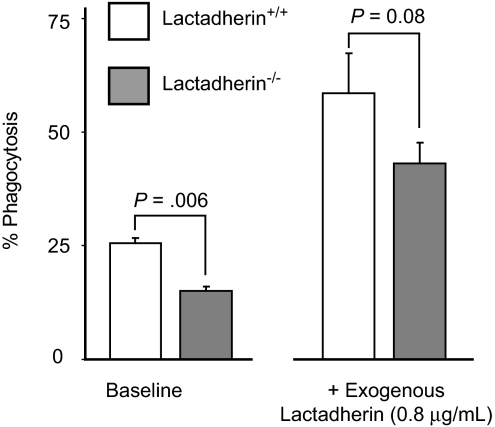
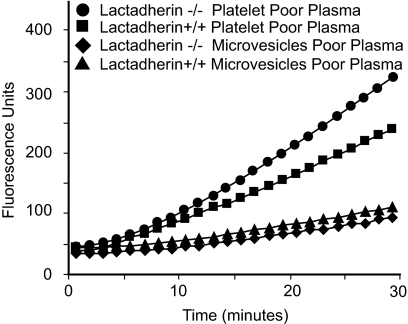
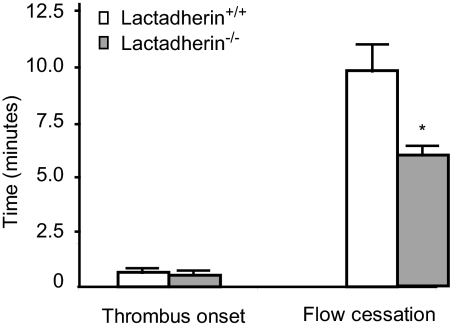
 ) and the littermate controls (□). *P = .01; n = 9.
) and the littermate controls (□). *P = .01; n = 9.References
-
- Bevers EM, Comfurius P, Zwaal RF. Regulatory mechanisms in maintenance and modulation of transmembrane lipid asymmetry: pathophysiological implications. Lupus. 1996;5:480–487. - PubMed
-
- Zwaal RF, Schroit AJ. Pathophysiologic implications of membrane phospholipid asymmetry in blood cells. Blood. 1997;89:1121–1132. - PubMed
-
- Solum NO. Procoagulant expression in platelets and defects leading to clinical disorders. Arterioscler Thromb Vasc Biol. 1999;19:2841–2846. - PubMed
-
- Comfurius P, Senden JM, Tilly RH, Schroit AJ, Bevers EM, Zwaal RF. Loss of membrane phospholipid asymmetry in platelets and red cells may be associated with calcium-induced shedding of plasma membrane and inhibition of aminophospholipid translocase. Biochim Biophys Acta. 1990;1026:153–160. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
