

Mutations in glucose transporter 9 gene SLC2A9 cause renal hypouricemia
- PMID: 19026395
- PMCID: PMC2668068
- DOI: 10.1016/j.ajhg.2008.11.001
Mutations in glucose transporter 9 gene SLC2A9 cause renal hypouricemia
Erratum in
- Am J Hum Genet. 2008 Dec;83(6):795
Abstract
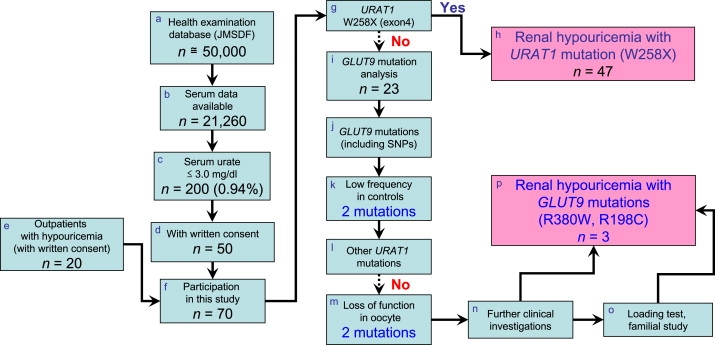
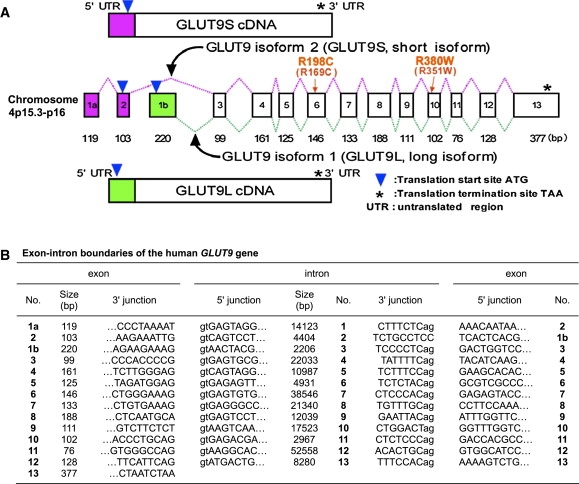
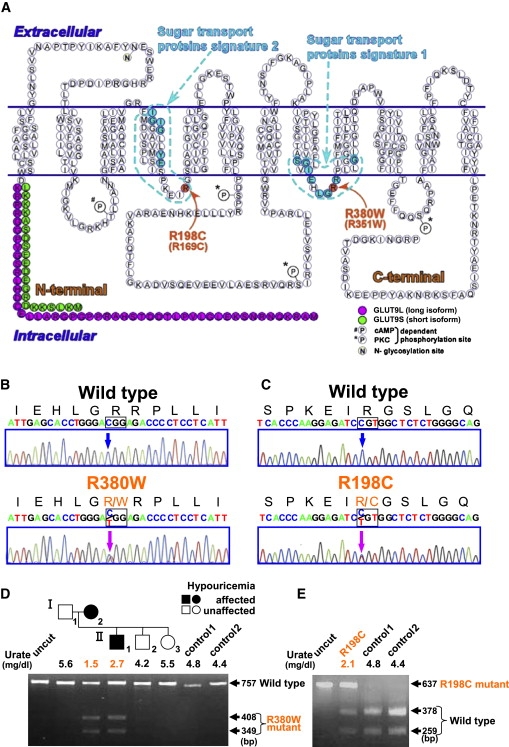
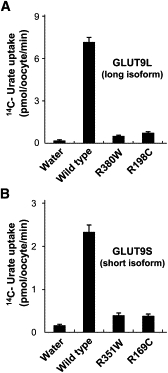
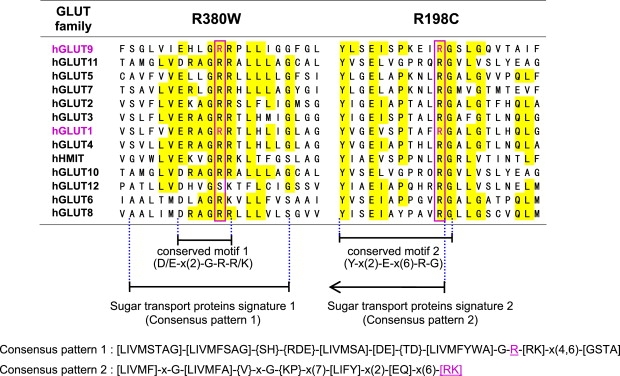
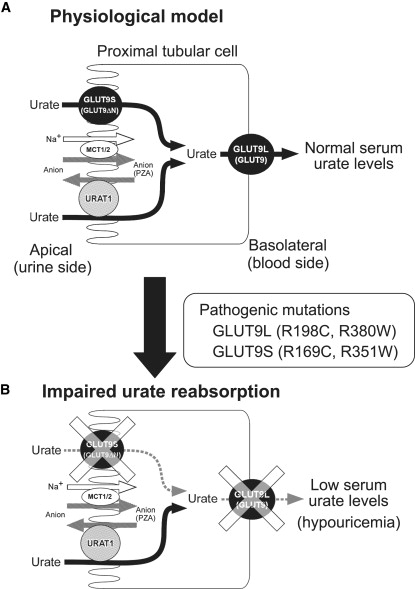
Renal hypouricemia is an inherited disorder characterized by impaired renal urate (uric acid) reabsorption and subsequent low serum urate levels, with severe complications such as exercise-induced acute renal failure and nephrolithiasis. We previously identified SLC22A12, also known as URAT1, as a causative gene of renal hypouricemia. However, hypouricemic patients without URAT1 mutations, as well as genome-wide association studies between urate and SLC2A9 (also called GLUT9), imply that GLUT9 could be another causative gene of renal hypouricemia. With a large human database, we identified two loss-of-function heterozygous mutations in GLUT9, which occur in the highly conserved "sugar transport proteins signatures 1/2." Both mutations result in loss of positive charges, one of which is reported to be an important membrane topology determinant. The oocyte expression study revealed that both GLUT9 isoforms showed high urate transport activities, whereas the mutated GLUT9 isoforms markedly reduced them. Our findings, together with previous reports on GLUT9 localization, suggest that these GLUT9 mutations cause renal hypouricemia by their decreased urate reabsorption on both sides of the renal proximal tubules. These findings also enable us to propose a physiological model of the renal urate reabsorption in which GLUT9 regulates serum urate levels in humans and can be a promising therapeutic target for gout and related cardiovascular diseases.
Figures
References
-
- Kikuchi Y., Koga H., Yasutomo Y., Kawabata Y., Shimizu E., Naruse M., Kiyama S., Nonoguchi H., Tomita K., Sasatomi Y. Patients with renal hypouricemia with exercise-induced acute renal failure and chronic renal dysfunction. Clin. Nephrol. 2000;53:467–472. - PubMed
-
- Enomoto A., Kimura H., Chairoungdua A., Shigeta Y., Jutabha P., Cha S.H., Hosoyamada M., Takeda M., Sekine T., Igarashi T. Molecular identification of a renal urate anion exchanger that regulates blood urate levels. Nature. 2002;417:447–452. - PubMed
-
- Wakida N., Tuyen D.G., Adachi M., Miyoshi T., Nonoguchi H., Oka T., Ueda O., Tazawa M., Kurihara S., Yoneta Y. Mutations in human urate transporter 1 gene in presecretory reabsorption defect type of familial renal hypouricemia. J. Clin. Endocrinol. Metab. 2005;90:2169–2174. - PubMed
-
- Ichida K., Hosoyamada M., Hisatome I., Enomoto A., Hikita M., Endou H., Hosoya T. Clinical and molecular analysis of patients with renal hypouricemia in Japan-influence of URAT1 gene on urinary urate excretion. J. Am. Soc. Nephrol. 2004;15:164–173. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
