

Notch and Kras reprogram pancreatic acinar cells to ductal intraepithelial neoplasia
- PMID: 19028876
- PMCID: PMC2585942
- DOI: 10.1073/pnas.0810111105
Notch and Kras reprogram pancreatic acinar cells to ductal intraepithelial neoplasia
Abstract
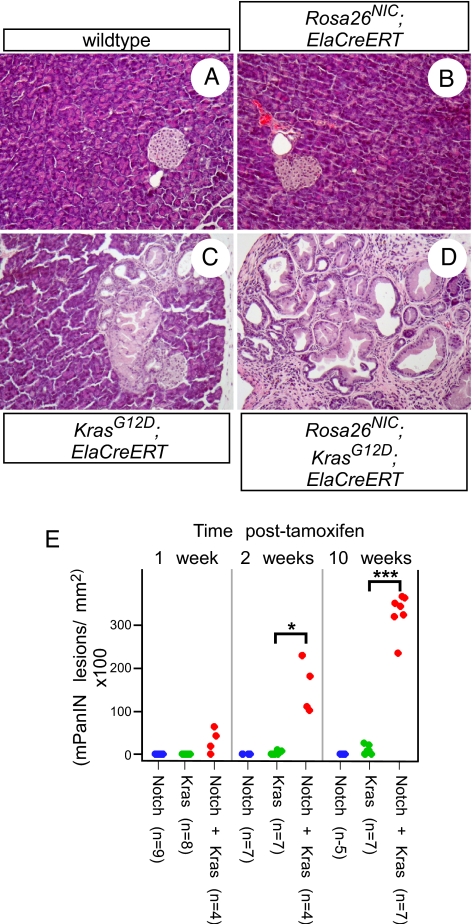
Efforts to model pancreatic cancer in mice have focused on mimicking genetic changes found in the human disease, particularly the activating KRAS mutations that occur in pancreatic tumors and their putative precursors, pancreatic intraepithelial neoplasia (PanIN). Although activated mouse Kras mutations induce PanIN lesions similar to those of human, only a small minority of cells that express mutant Kras go on to form PanINs. The basis for this selective response is unknown, and it is similarly unknown what cell types in the mature pancreas actually contribute to PanINs. One clue comes from the fact that PanINs, unlike most cells in the adult pancreas, exhibit active Notch signaling. We hypothesize that Notch, which inhibits differentiation in the embryonic pancreas, contributes to PanIN formation by abrogating the normal differentiation program of tumor-initiating cells. Through conditional expression in the mouse pancreas, we find dramatic synergy between activated Notch and Kras in inducing PanIN formation. Furthermore, we find that Kras activation in mature acinar cells induces PanIN lesions identical to those seen upon ubiquitous Kras activation, and that Notch promotes both initiation and dysplastic progression of these acinar-derived PanINs, albeit short of invasive adenocarcinoma. At the cellular level, Notch/Kras coactivation promotes rapid reprogramming of acinar cells to a duct-like phenotype, providing an explanation for how a characteristically ductal tumor can arise from nonductal acinar cells.
Conflict of interest statement
The authors declare no conflict of interest.
Figures



References
-
- Hingorani SR, et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell. 2003;4:437–450. - PubMed
-
- Hruban RH, et al. Pathology of genetically engineered mouse models of pancreatic exocrine cancer: Consensus report and recommendations. Cancer Res. 2006;66:95–106. - PubMed
-
- Jensen J. Gene regulatory factors in pancreatic development. Dev Dyn. 2004;229:176–200. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
