

Roles of IP3R and RyR Ca2+ channels in endoplasmic reticulum stress and beta-cell death
- PMID: 19033399
- PMCID: PMC2628616
- DOI: 10.2337/db07-1762
Roles of IP3R and RyR Ca2+ channels in endoplasmic reticulum stress and beta-cell death
Abstract
Objective: Endoplasmic reticulum (ER) stress has been implicated in the pathogenesis of diabetes, but the roles of specific ER Ca(2+) release channels in the ER stress-associated apoptosis pathway remain unknown. Here, we examined the effects of stimulating or inhibiting the ER-resident inositol trisphosphate receptors (IP(3)Rs) and the ryanodine receptors (RyRs) on the induction of beta-cell ER stress and apoptosis.
Research design and methods: Kinetics of beta-cell death were tracked by imaging propidium iodide incorporation and caspase-3 activity in real time. ER stress and apoptosis were assessed by Western blot. Mitochondrial membrane potential was monitored by flow cytometry. Cytosolic Ca(2+) was imaged using fura-2, and genetically encoded fluorescence resonance energy transfer (FRET)-based probes were used to measure Ca(2+) in ER and mitochondria.
Results: Neither RyR nor IP(3)R inhibition, alone or in combination, caused robust death within 24 h. In contrast, blocking sarco/endoplasmic reticulum ATPase (SERCA) pumps depleted ER Ca(2+) and induced marked phosphorylation of PKR-like ER kinase (PERK) and eukaryotic initiation factor-2alpha (eIF2alpha), C/EBP homologous protein (CHOP)-associated ER stress, caspase-3 activation, and death. Notably, ER stress following SERCA inhibition was attenuated by blocking IP(3)Rs and RyRs. Conversely, stimulation of ER Ca(2+) release channels accelerated thapsigargin-induced ER depletion and apoptosis. SERCA block also activated caspase-9 and induced perturbations of the mitochondrial membrane potential, resulting eventually in the loss of mitochondrial polarization.
Conclusions: This study demonstrates that the activity of ER Ca(2+) channels regulates the susceptibility of beta-cells to ER stress resulting from impaired SERCA function. Our results also suggest the involvement of mitochondria in beta-cell apoptosis associated with dysfunctional beta-cell ER Ca(2+) homeostasis and ER stress.
Figures

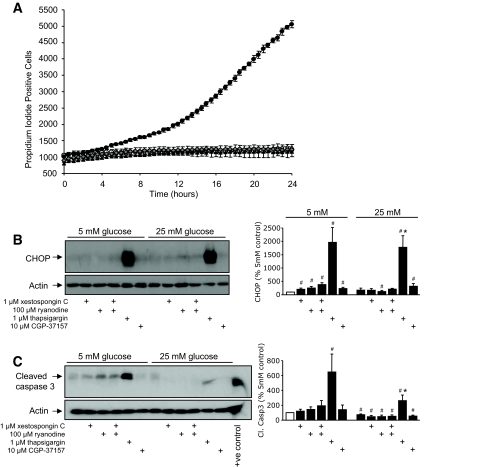
 , 100 μmol/l ryanodine + 1 μmol/l xestaspongin C; •, 1 μmol/l thapsigargin. B: MIN6 cells were cultured as indicated and probed for markers of ER stress and apoptosis, as in Fig. 2. CHOP expression was examined and quantified at both low and high glucose (n = 4–10). C: Cleaved caspase-3 expression examined and quantified at both low and high glucose (n = 4–10). A positive control for cleaved (Cl.) caspase-3 supplied by the manufacturer (lysates from apoptotic T-cells) was included in the final lane. #P < 0.05 vs. 5 mmol/l glucose control; *P < 0.05 vs. 25 mmol/l glucose control.
, 100 μmol/l ryanodine + 1 μmol/l xestaspongin C; •, 1 μmol/l thapsigargin. B: MIN6 cells were cultured as indicated and probed for markers of ER stress and apoptosis, as in Fig. 2. CHOP expression was examined and quantified at both low and high glucose (n = 4–10). C: Cleaved caspase-3 expression examined and quantified at both low and high glucose (n = 4–10). A positive control for cleaved (Cl.) caspase-3 supplied by the manufacturer (lysates from apoptotic T-cells) was included in the final lane. #P < 0.05 vs. 5 mmol/l glucose control; *P < 0.05 vs. 25 mmol/l glucose control. , 1 μmol/l thapsigargin + 30 μmol/l dantrolene. A trend toward protection from thapsigargin-induced death was also observed in response to treatment with 100 μmol/l ryanodine alone (n = 4) or in combination with 1 μmol/l xestospongin C (n = 6). C and D: Caspase-3 activation following an 8-h treatment with thapsigargin was reduced by dantrolene (n = 3). E: Caspase-3 cleavage in MIN6 cells cultured at 5 mmol/l glucose for 24 h with 1 μmol/l thapsigargin in the presence or absence of both 100 μmol/l ryanodine and 1 μmol/l xestospongin C (n = 6). F and G: Quantified Western blots of cleaved caspase-3 and CHOP levels in MIN6 cells treated for 8 h in 5 mmol/l glucose as indicated (n = 3). #P < 0.05 vs. control; *P < 0.05 vs. thapsigargin alone.
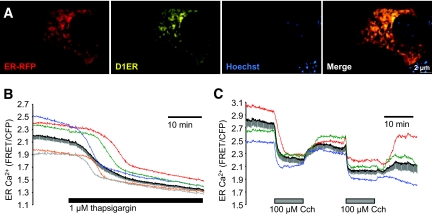
, 1 μmol/l thapsigargin + 30 μmol/l dantrolene. A trend toward protection from thapsigargin-induced death was also observed in response to treatment with 100 μmol/l ryanodine alone (n = 4) or in combination with 1 μmol/l xestospongin C (n = 6). C and D: Caspase-3 activation following an 8-h treatment with thapsigargin was reduced by dantrolene (n = 3). E: Caspase-3 cleavage in MIN6 cells cultured at 5 mmol/l glucose for 24 h with 1 μmol/l thapsigargin in the presence or absence of both 100 μmol/l ryanodine and 1 μmol/l xestospongin C (n = 6). F and G: Quantified Western blots of cleaved caspase-3 and CHOP levels in MIN6 cells treated for 8 h in 5 mmol/l glucose as indicated (n = 3). #P < 0.05 vs. control; *P < 0.05 vs. thapsigargin alone. , 1 μmol/l thapsigargin + 100 μmol/l Cch. B: ○, control; •, 1 μmol/l thapsigargin; , 1 μmol/l thapsigargin + 1 nmol/l ryanodine. D and E: MIN6 cells cultured for 8 h with a combination of 100 μmol/l carbachol (Cch) and 1 μmol/l thapsigargin showed increased ER stress (CHOP expression), compared with either treatment alone (n = 6). #P < 0.05 vs. control; *P < 0.05 vs. thapsigargin alone.
, 1 μmol/l thapsigargin + 100 μmol/l Cch. B: ○, control; •, 1 μmol/l thapsigargin; , 1 μmol/l thapsigargin + 1 nmol/l ryanodine. D and E: MIN6 cells cultured for 8 h with a combination of 100 μmol/l carbachol (Cch) and 1 μmol/l thapsigargin showed increased ER stress (CHOP expression), compared with either treatment alone (n = 6). #P < 0.05 vs. control; *P < 0.05 vs. thapsigargin alone.
 , 100 μmol/l Cch; ▪, 1 μmol/l thapsigargin;
, 100 μmol/l Cch; ▪, 1 μmol/l thapsigargin;  , thapsigargin + Cch. (Please see
, thapsigargin + Cch. (Please see 

References
-
- Harding HP, Ron D: Endoplasmic reticulum stress and the development of diabetes: a review. Diabetes 51 (Suppl. 3): S455–S461, 2002 - PubMed
-
- Huang CJ, Lin CY, Haataja L, Gurlo T, Butler AE, Rizza RA, Butler PC: High expression rates of human islet amyloid polypeptide induce endoplasmic reticulum stress–mediated β-cell apoptosis, a characteristic of humans with type 2 but not type 1 diabetes. Diabetes 56: 2016–2027, 2007 - PubMed
-
- Cnop M, Welsh N, Jonas JC, Jorns A, Lenzen S, Eizirik DL: Mechanisms of pancreatic β-cell death in type 1 and type 2 diabetes: many differences, few similarities. Diabetes 54 (Suppl. 2): S97–S107, 2005 - PubMed
-
- Yamada T, Ishihara H, Tamura A, Takahashi R, Yamaguchi S, Takei D, Tokita A, Satake C, Tashiro F, Katagiri H, Aburatani H, Miyazaki J, Oka Y: WFS1-deficiency increases endoplasmic reticulum stress, impairs cell cycle progression and triggers the apoptotic pathway specifically in pancreatic β-cells. Hum Mol Genet 15: 1600–1609, 2006 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
