

Human immunodeficiency virus protein Tat induces synapse loss via a reversible process that is distinct from cell death
- PMID: 19036954
- PMCID: PMC2678679
- DOI: 10.1523/JNEUROSCI.2958-08.2008
Human immunodeficiency virus protein Tat induces synapse loss via a reversible process that is distinct from cell death
Abstract
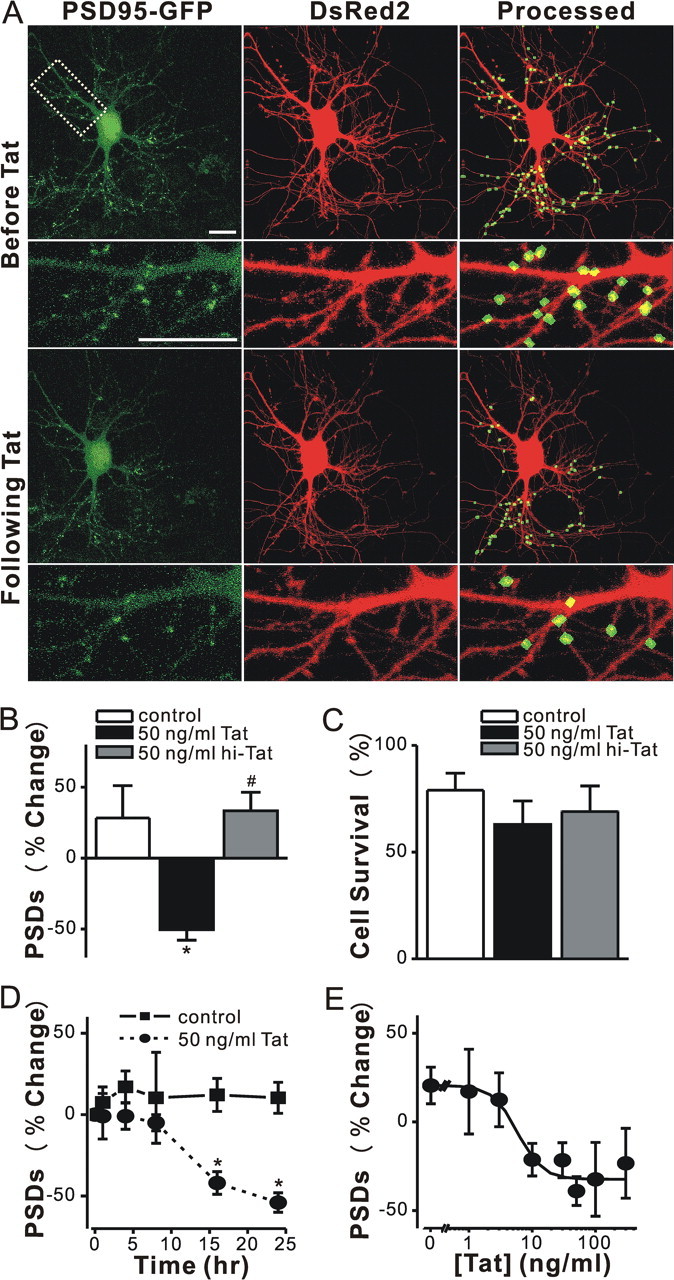
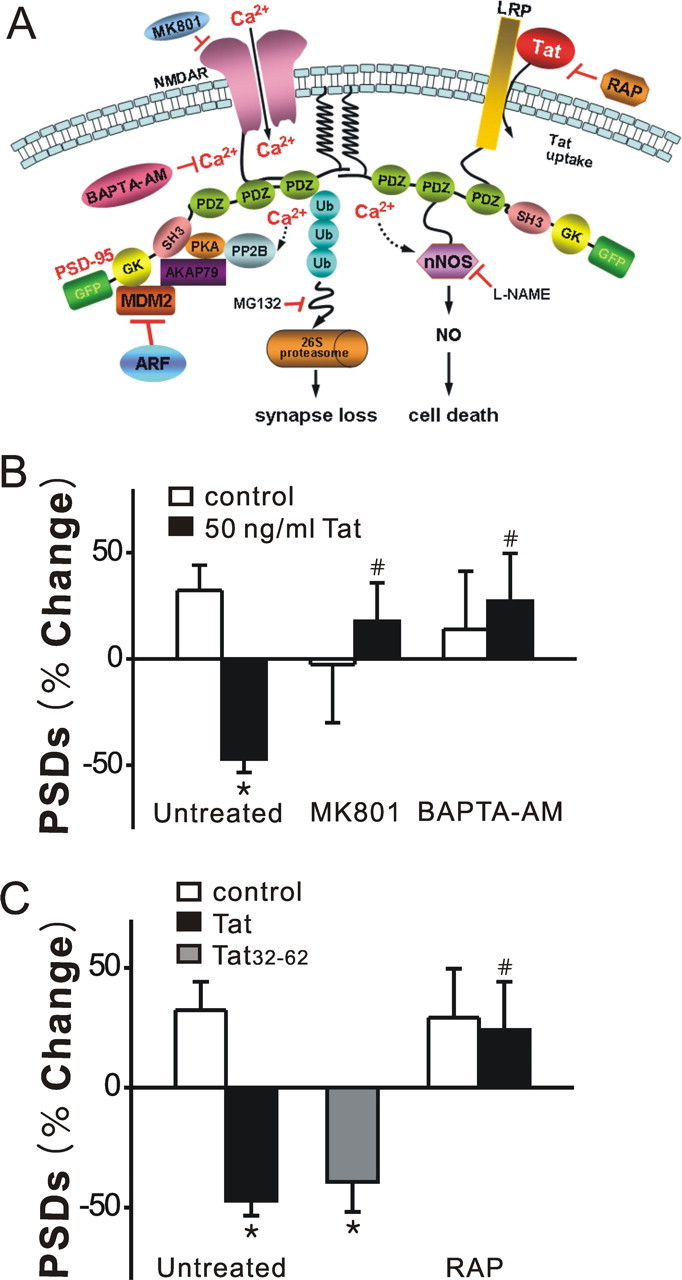
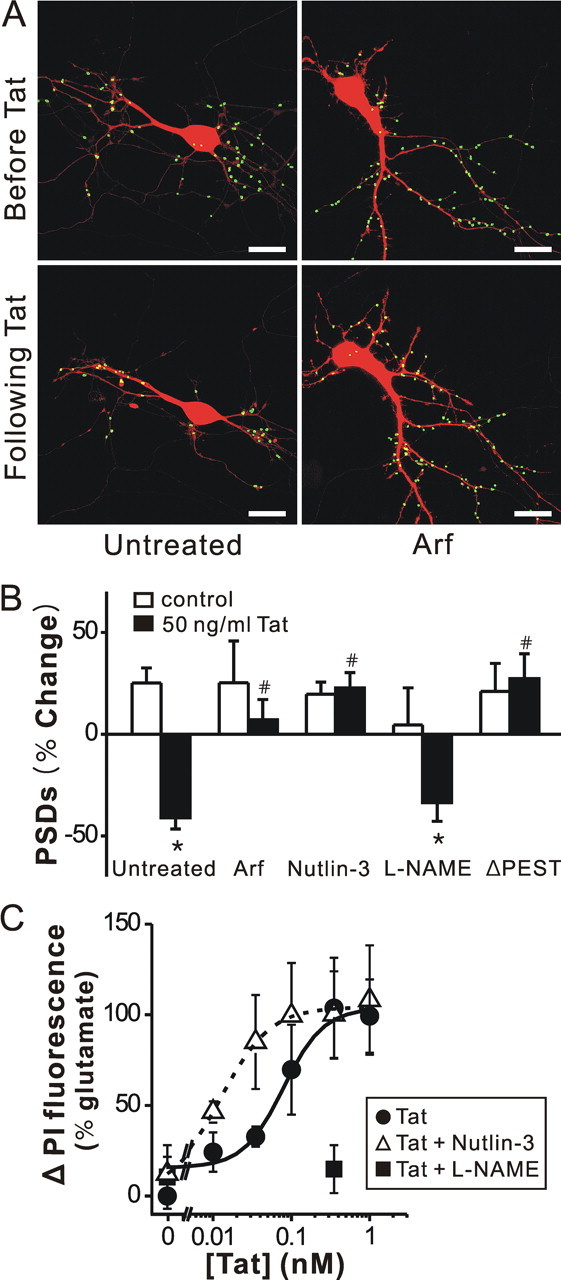
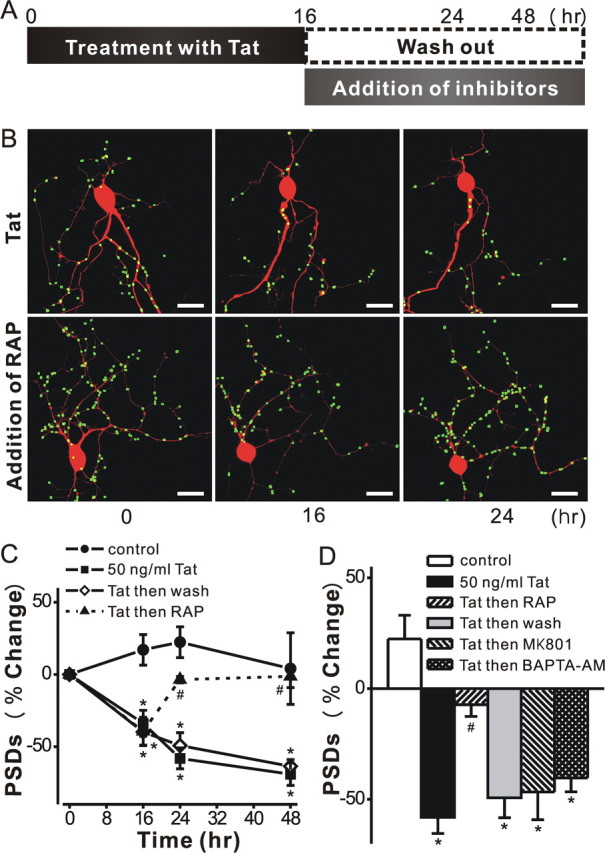
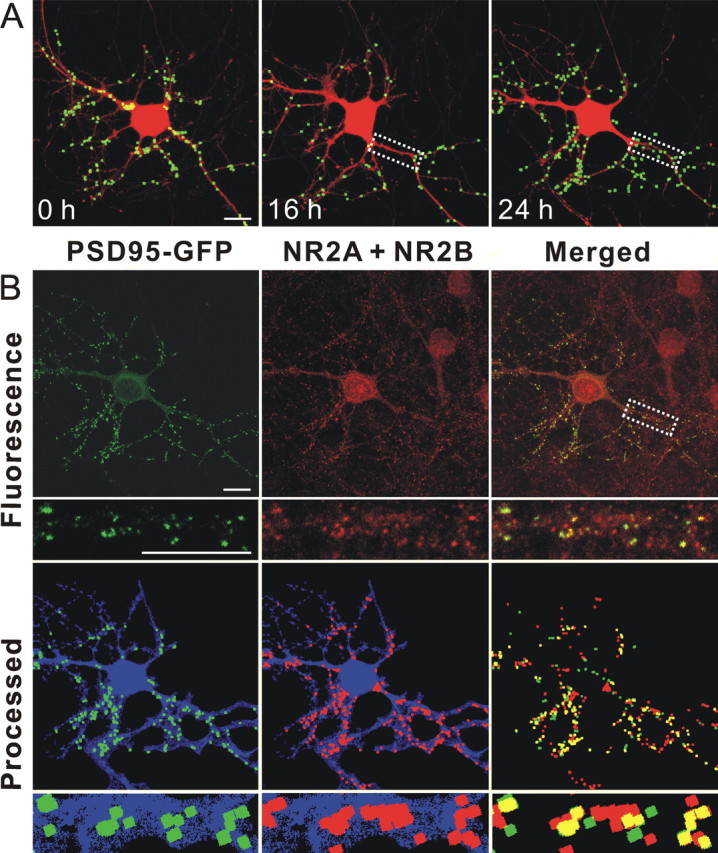
Human immunodeficiency virus (HIV)-1 infection of the CNS produces changes in dendritic morphology that correlate with cognitive decline in patients with HIV-1 associated dementia (HAD). Here, we investigated the effects of HIV-1 transactivator of transcription (Tat), a protein released by virus-infected cells, on synapses between hippocampal neurons using an imaging-based assay that quantified clusters of the scaffolding protein postsynaptic density 95 fused to green fluorescent protein (PSD95-GFP). Tat (24 h) decreased the number of PSD95-GFP puncta by 50 +/- 7%. The decrease was concentration-dependent (EC(50) = 6 +/- 2 ng/ml) and preceded cell death. Tat acted via the low-density lipoprotein receptor-related protein (LRP) because the specific LRP blocker, receptor associated protein (RAP), prevented the Tat-induced decrease in the number of PSD95-GFP puncta. Ca(2+) influx through the NMDA receptor was necessary for Tat-induced synapse loss. Expression of an ubiquitin ligase inhibitor protected synapses, implicating the ubiquitin-proteasome pathway. In contrast to synapse loss, Tat induced cell death (48 h) required activation of nitric oxide synthase. The ubiquitin ligase-inhibitor nutlin-3 prevented synapse loss but not cell death induced by Tat. Thus, the pathways diverged, consistent with the hypothesis that synapse loss is a mechanism to reduce excess excitatory input rather than a symptom of the neuron's demise. Furthermore, application of RAP to cultures treated with Tat for 16 h reversed synapse loss. These results suggest that the impaired network function and decreased neuronal survival produced by Tat involve distinct mechanisms and that pharmacologic targets, such as LRP, might prove useful in restoring function in HAD patients.
Figures
References
-
- Adle-Biassette H, Chrétien F, Wingertsmann L, Héry C, Ereau T, Scaravilli F, Tardieu M, Gray F. Neuronal apoptosis does not correlate with dementia in HIV infection but is related to microglial activation and axonal damage. Neuropathol Appl Neurobiol. 1999;25:123–133. - PubMed
-
- Albensi BC. The NMDA receptor/ion channel complex: a drug target for modulating synaptic plasticity and excitotoxicity. Curr Pharm Des. 2007;13:3185–3194. - PubMed
-
- Arnold DB, Clapham DE. Molecular determinants for subcellular localization of PSD-95 with an interacting K+ channel. Neuron. 1999;23:149–157. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous