

Oxidative-stress-induced afterdepolarizations and calmodulin kinase II signaling
- PMID: 19038865
- PMCID: PMC2747806
- DOI: 10.1161/CIRCRESAHA.108.183475
Oxidative-stress-induced afterdepolarizations and calmodulin kinase II signaling
Abstract
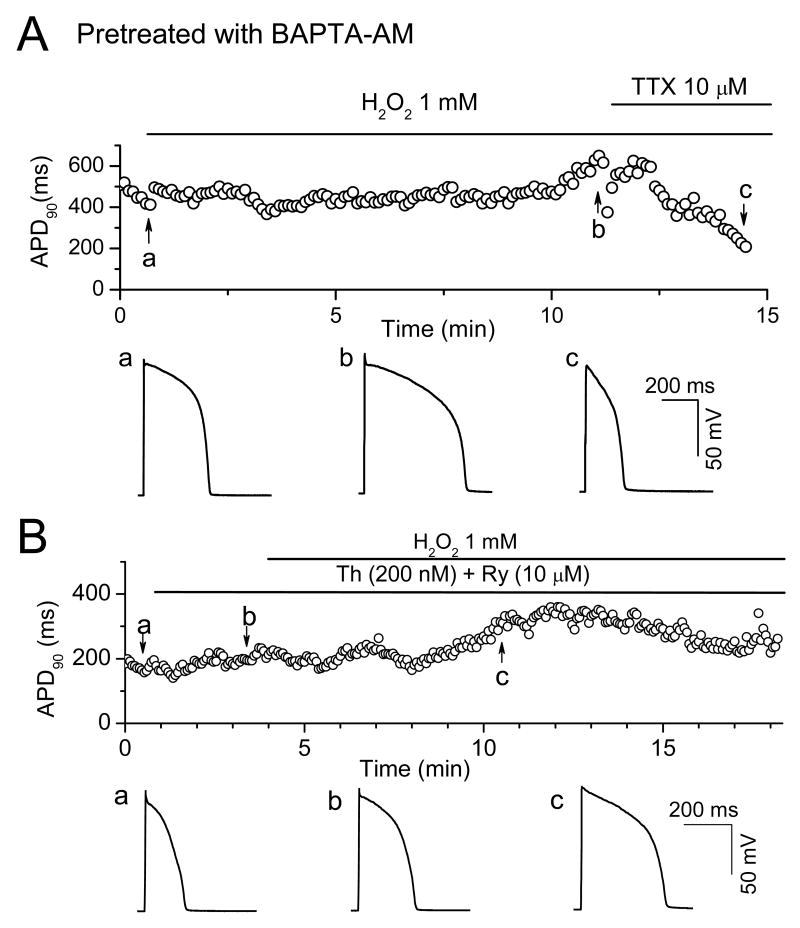
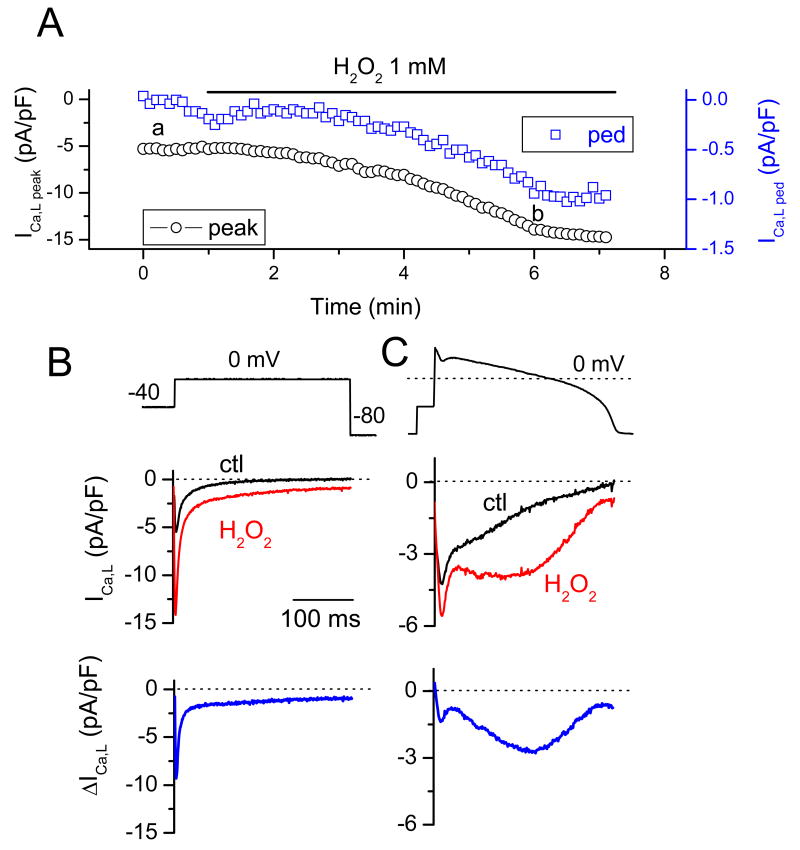
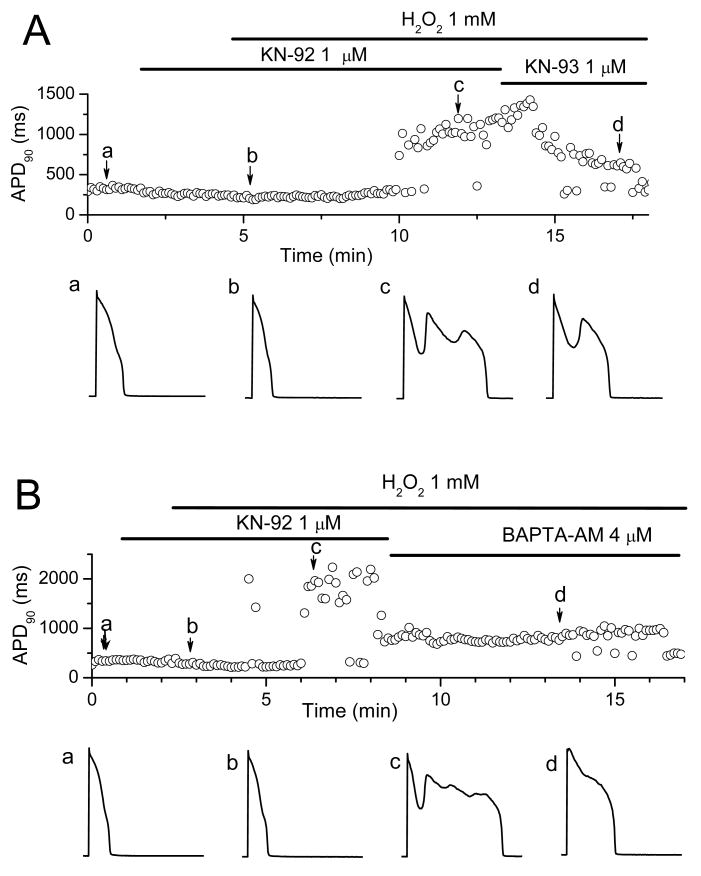
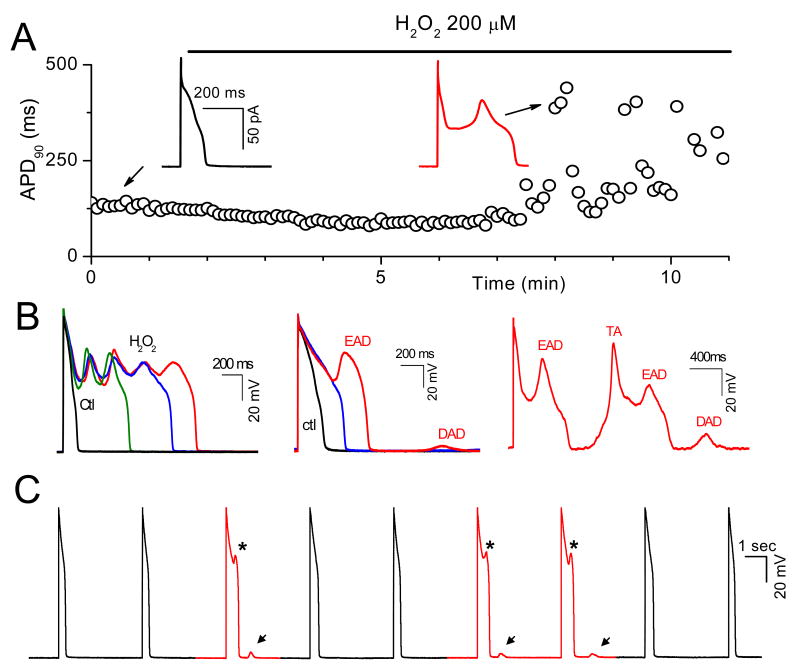
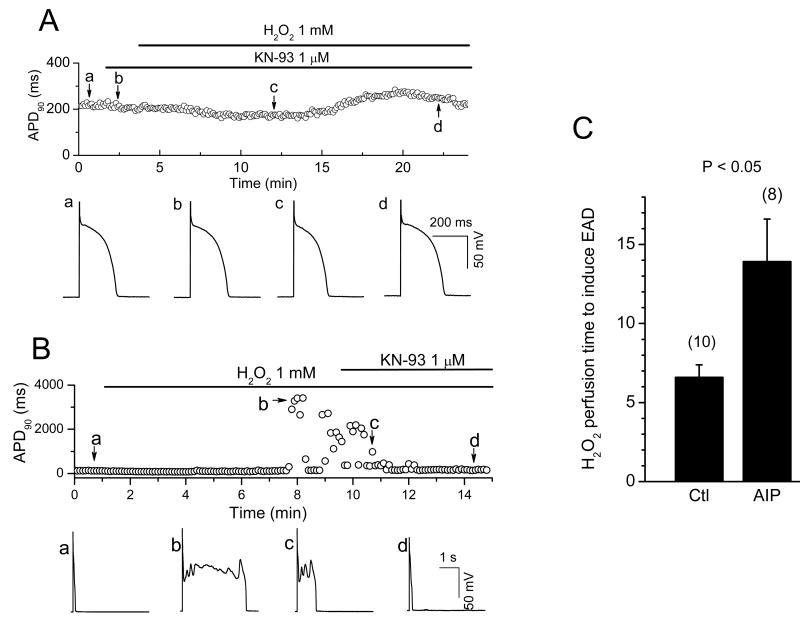
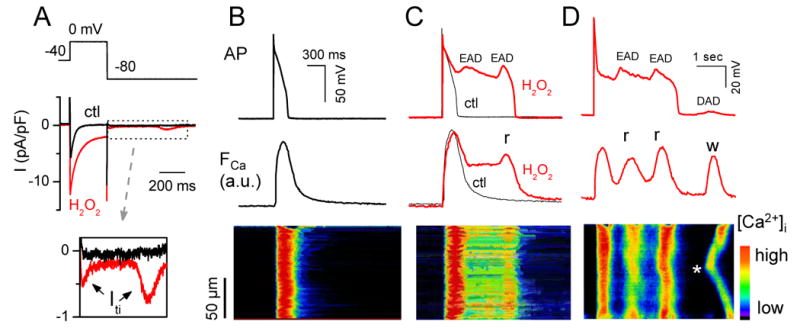
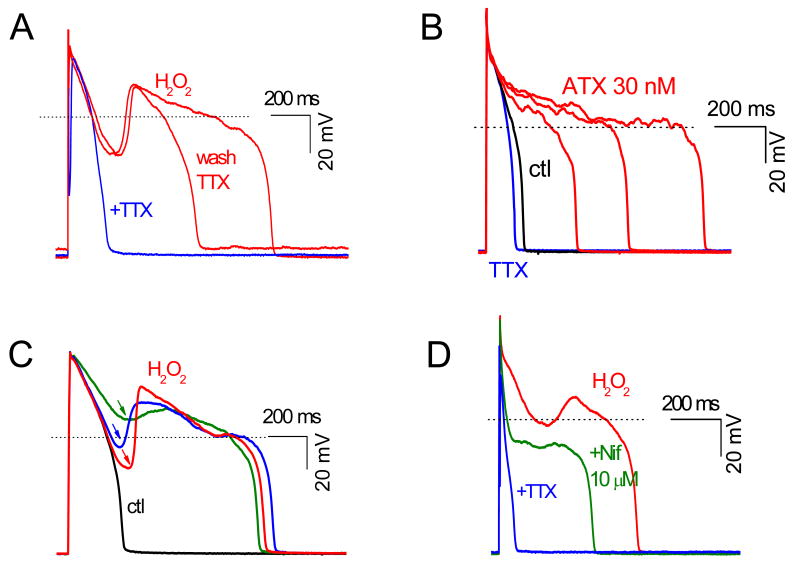
In the heart, oxidative stress caused by exogenous H(2)O(2) has been shown to induce early afterdepolarizations (EADs) and triggered activity by impairing Na current (I(Na)) inactivation. Because H(2)O(2) activates Ca(2+)/calmodulin kinase (CaMK)II, which also impairs I(Na) inactivation and promotes EADs, we hypothesized that CaMKII activation may be an important factor in EADs caused by oxidative stress. Using the patch-clamp and intracellular Ca (Ca(i)) imaging in Fluo-4 AM-loaded rabbit ventricular myocytes, we found that exposure to H(2)O(2) (0.2 to 1 mmol/L) for 5 to 15 minutes consistently induced EADs that were suppressed by the I(Na) blocker tetrodotoxin (10 micromol/L), as well as the I(Ca,L) blocker nifedipine. H(2)O(2) enhanced both peak and late I(Ca,L), consistent with CaMKII-mediated facilitation. By prolonging the action potential plateau and increasing Ca influx via I(Ca,L), H(2)O(2)-induced EADs were also frequently followed by DADs in response to spontaneous (ie, non-I(Ca,L)-gated) sarcoplasmic reticulum Ca release after repolarization. The CaMKII inhibitor KN-93 (1 micromol/L; n=4), but not its inactive analog KN-92 (1 micromol/L, n=5), prevented H(2)O(2)-induced EADs and DADs, and the selective CaMKII peptide inhibitor AIP (autocamtide-2-related inhibitory peptide) (2 micromol/L) significantly delayed their onset. In conclusion, H(2)O(2)-induced afterdepolarizations depend on both impaired I(Na) inactivation to reduce repolarization reserve and enhancement of I(Ca,L) to reverse repolarization, which are both facilitated by CaMKII activation. Our observations support a link between increased oxidative stress, CaMKII activation, and afterdepolarizations as triggers of lethal ventricular arrhythmias in diseased hearts.
Conflict of interest statement
Figures
References
-
- Lakatta EG, Sollott SJ. The “heartbreak” of older age. Mol Interv. 2002;2:431–446. - PubMed
-
- Song Y, Shryock JC, Wagner S, Maier LS, Belardinelli L. Blocking late sodium current reduces hydrogen peroxide-induced arrhythmogenic activity and contractile dysfunction. J Pharmacol Exp Ther. 2006;318:214–222. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
