

MG53 nucleates assembly of cell membrane repair machinery
- PMID: 19043407
- PMCID: PMC2990407
- DOI: 10.1038/ncb1812
MG53 nucleates assembly of cell membrane repair machinery
Abstract
Dynamic membrane repair and remodelling is an elemental process that maintains cell integrity and mediates efficient cellular function. Here we report that MG53, a muscle-specific tripartite motif family protein (TRIM72), is a component of the sarcolemmal membrane-repair machinery. MG53 interacts with phosphatidylserine to associate with intracellular vesicles that traffic to and fuse with sarcolemmal membranes. Mice null for MG53 show progressive myopathy and reduced exercise capability, associated with defective membrane-repair capacity. Injury of the sarcolemmal membrane leads to entry of the extracellular oxidative environment and MG53 oligomerization, resulting in recruitment of MG53-containing vesicles to the injury site. After vesicle translocation, entry of extracellular Ca(2+) facilitates vesicle fusion to reseal the membrane. Our data indicate that intracellular vesicle translocation and Ca(2+)-dependent membrane fusion are distinct steps involved in the repair of membrane damage and that MG53 may initiate the assembly of the membrane repair machinery in an oxidation-dependent manner.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
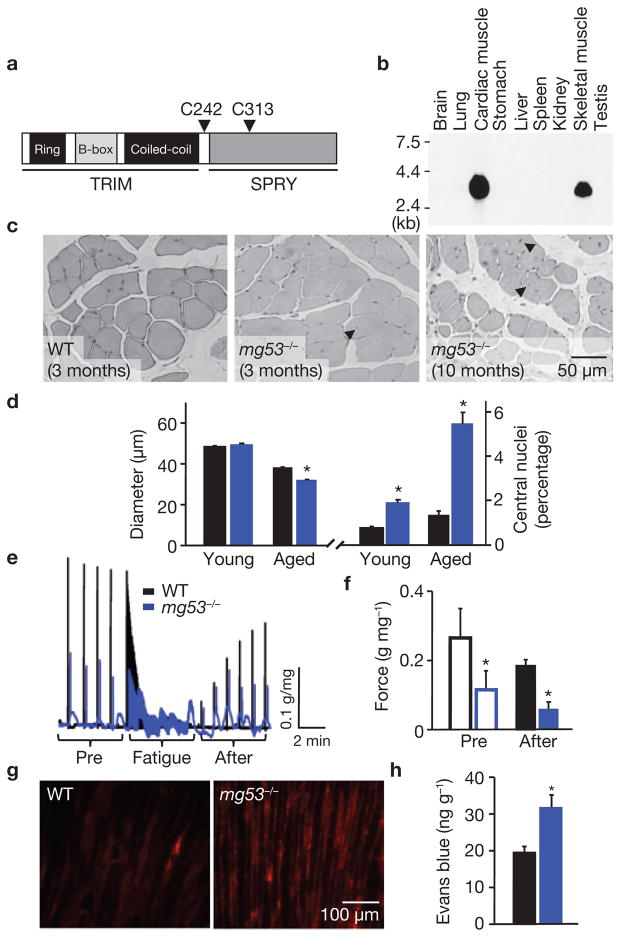
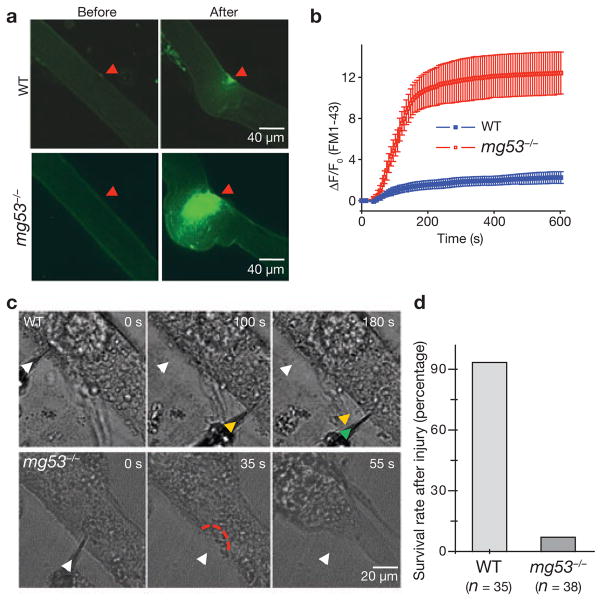
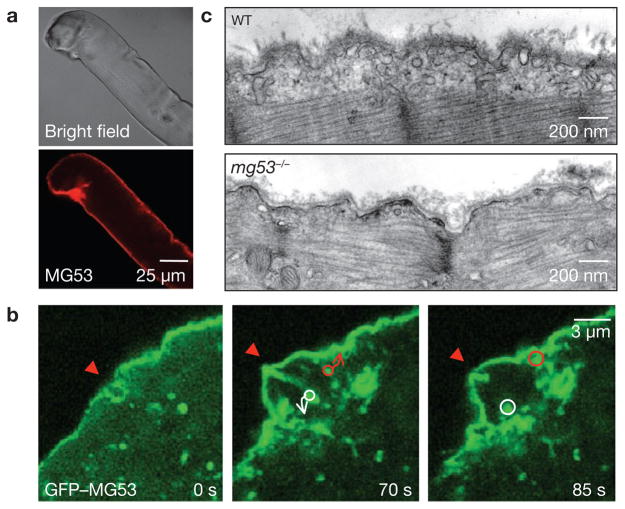
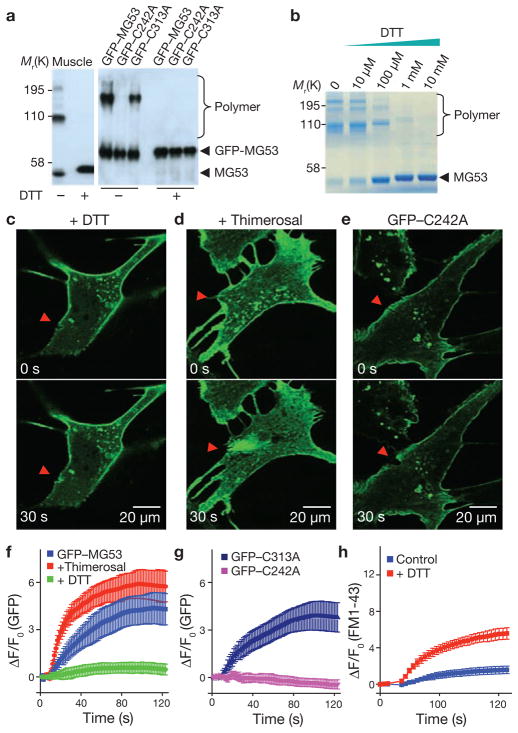
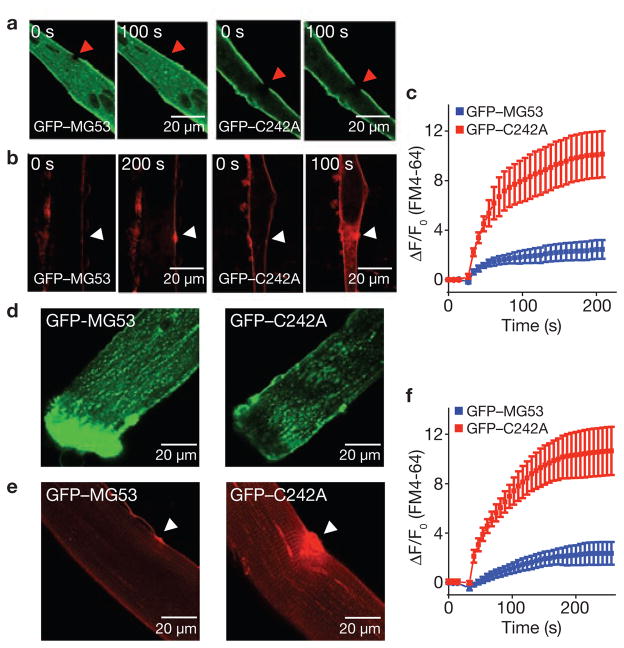
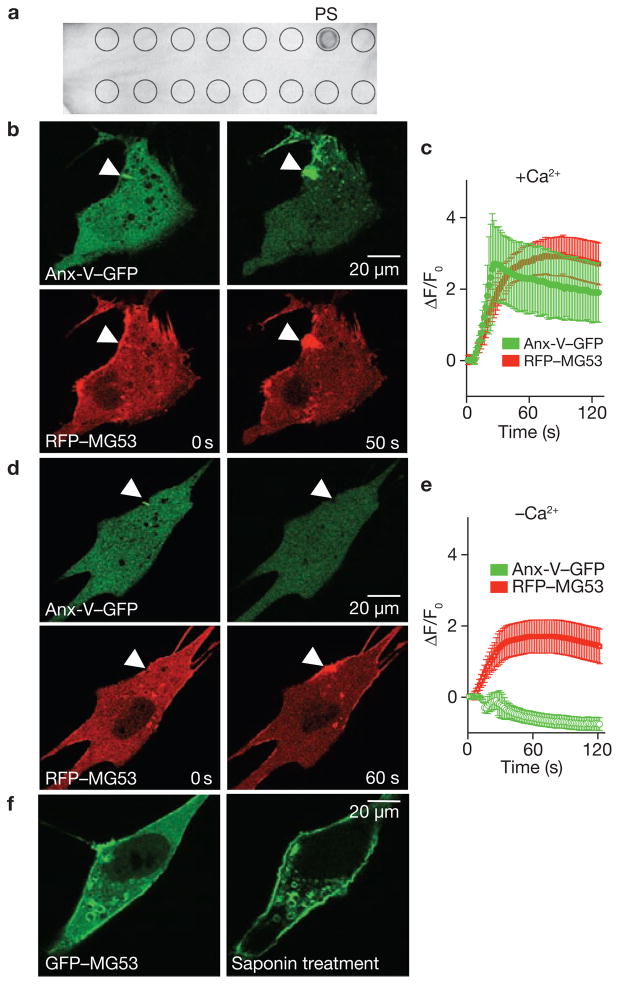
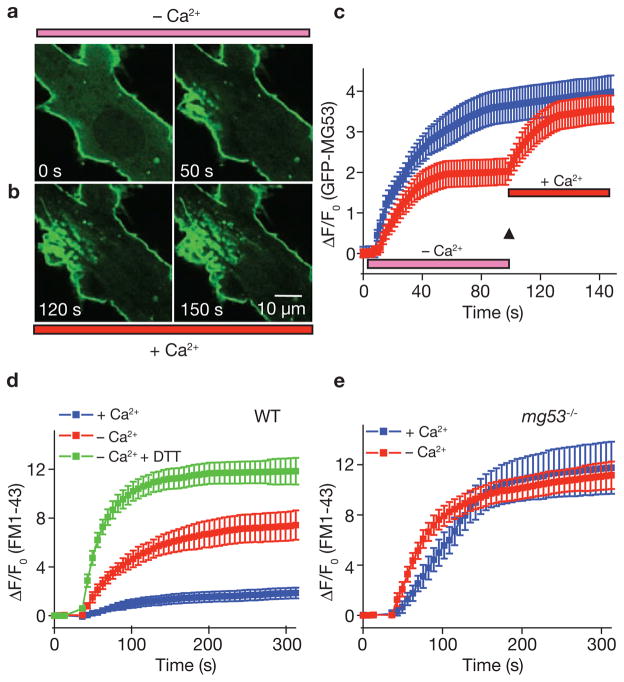
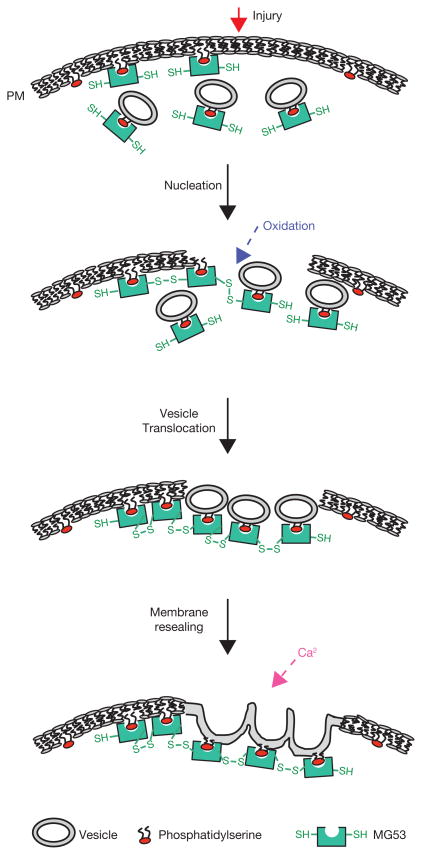
Comment in
-
Membrane repair redux: redox of MG53.Nat Cell Biol. 2009 Jan;11(1):7-9. doi: 10.1038/ncb0109-7. Nat Cell Biol. 2009. PMID: 19122591 No abstract available.
References
-
- Doherty KR, McNally EM. Repairing the tears: dysferlin in muscle membrane repair. Trends Mol Med. 2003;9:327–330. - PubMed
-
- Bansal D, Campbell KP. Dysferlin and the plasma membrane repair in muscular dystrophy. Trends Cell Biol. 2004;14:206–213. - PubMed
-
- Bazan NG, Marcheselli VL, Cole-Edwards K. Brain response to injury and neurodegeneration: endogenous neuroprotective signaling. Ann NY Acad Sci. 2005;1053:137–147. - PubMed
-
- McNeil PL, Kirchhausen T. An emergency response team for membrane repair. Nature Rev Mol Cell Biol. 2005;6:499–505. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
