

High-resolution mapping of copy-number alterations with massively parallel sequencing
- PMID: 19043412
- PMCID: PMC2630795
- DOI: 10.1038/nmeth.1276
High-resolution mapping of copy-number alterations with massively parallel sequencing
Abstract
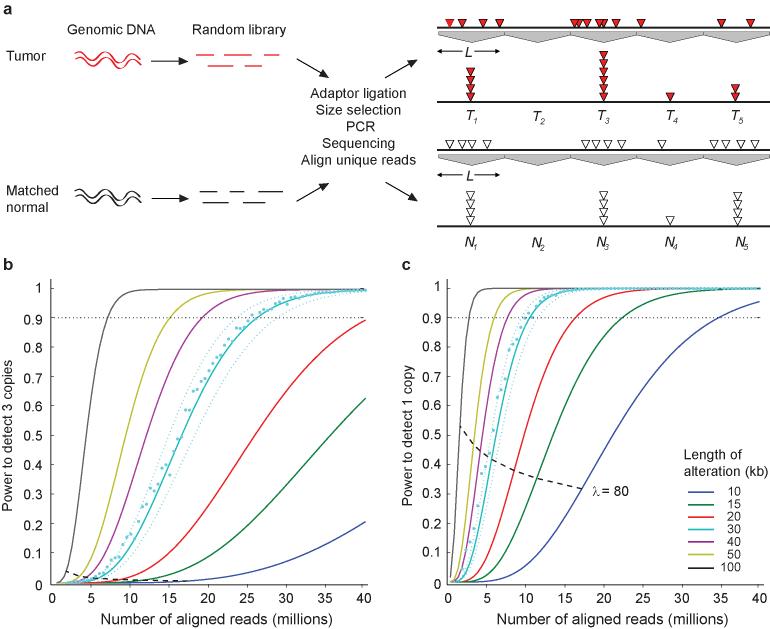
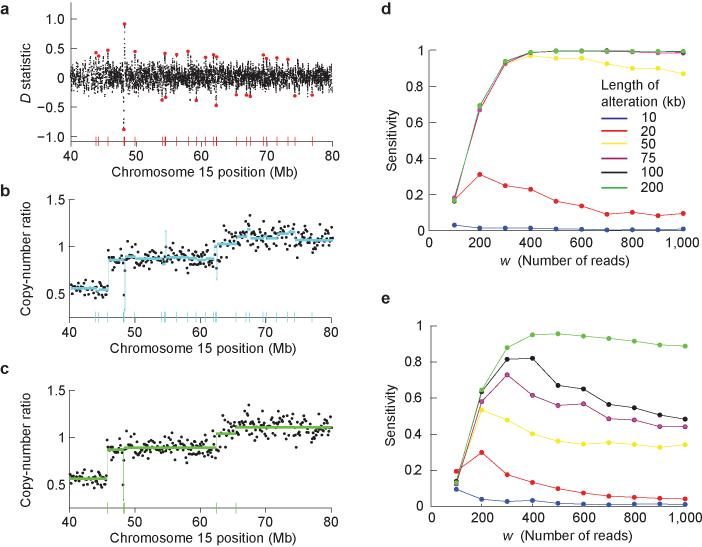
Cancer results from somatic alterations in key genes, including point mutations, copy-number alterations and structural rearrangements. A powerful way to discover cancer-causing genes is to identify genomic regions that show recurrent copy-number alterations (gains and losses) in tumor genomes. Recent advances in sequencing technologies suggest that massively parallel sequencing may provide a feasible alternative to DNA microarrays for detecting copy-number alterations. Here we present: (i) a statistical analysis of the power to detect copy-number alterations of a given size; (ii) SegSeq, an algorithm to segment equal copy numbers from massively parallel sequence data; and (iii) analysis of experimental data from three matched pairs of tumor and normal cell lines. We show that a collection of approximately 14 million aligned sequence reads from human cell lines has comparable power to detect events as the current generation of DNA microarrays and has over twofold better precision for localizing breakpoints (typically, to within approximately 1 kilobase).
Figures

References
-
- Freeman JL, et al. Copy number variation: New insights in genome diversity. Genome Res. 2006;16:949–961. - PubMed
-
- McCarroll SA, Altshuler DM. Copy-number variation and association studies of human disease. Nat. Genet. 2007;39:S37–S42. - PubMed
-
- Beckmann JS, Estivill X, Antonarakis SE. Copy number variants and genetic traits: closer to the resolution of phenotypic to genotypic variability. Nat. Rev. Genet. 2007;8:639–646. - PubMed
-
- Pinkel D, Albertson DG. Array comparative genomic hybridization and its applications in cancer. Nat. Genet. 2005;37:S11–S17. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
