

Genetic analysis of human traits in vitro: drug response and gene expression in lymphoblastoid cell lines
- PMID: 19043577
- PMCID: PMC2583954
- DOI: 10.1371/journal.pgen.1000287
Genetic analysis of human traits in vitro: drug response and gene expression in lymphoblastoid cell lines
Abstract
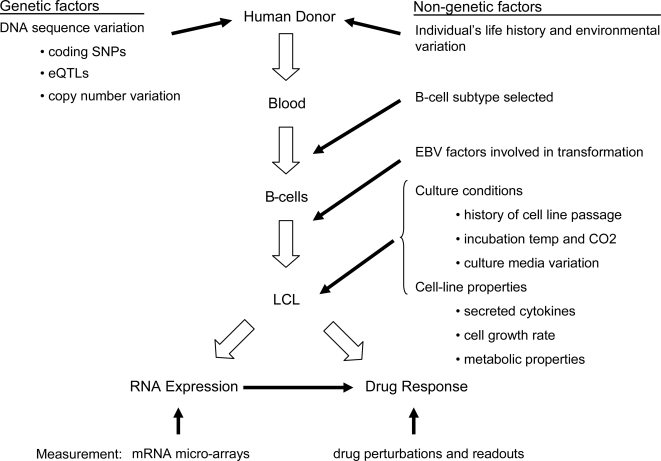
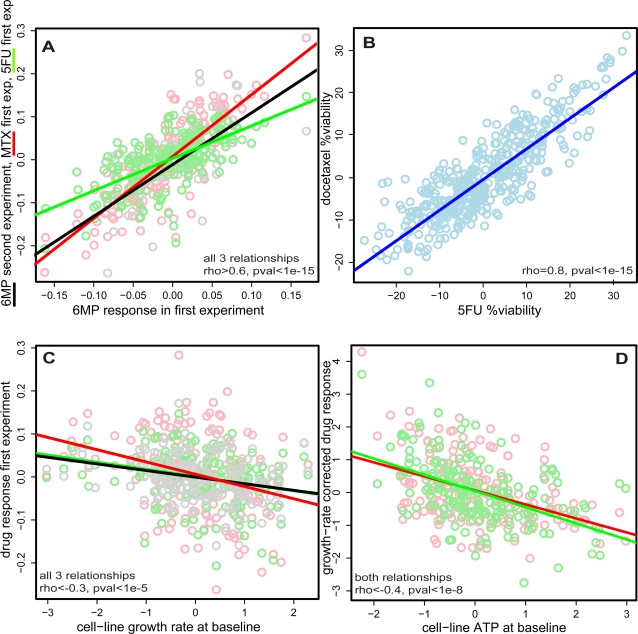
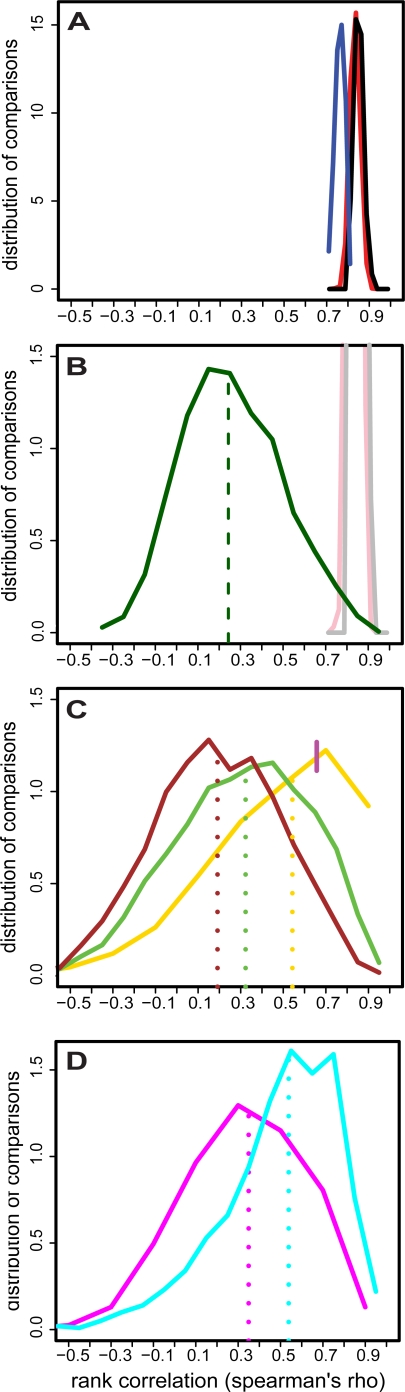
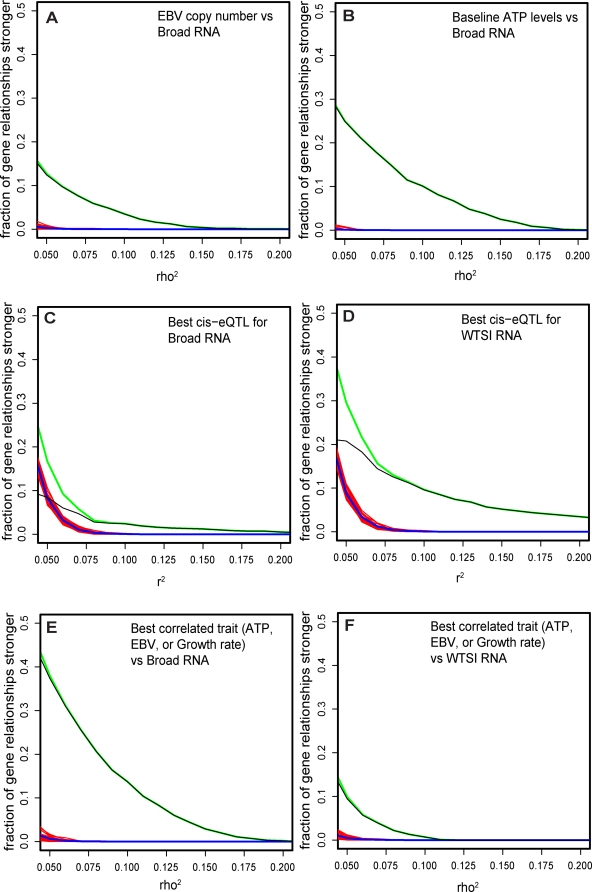
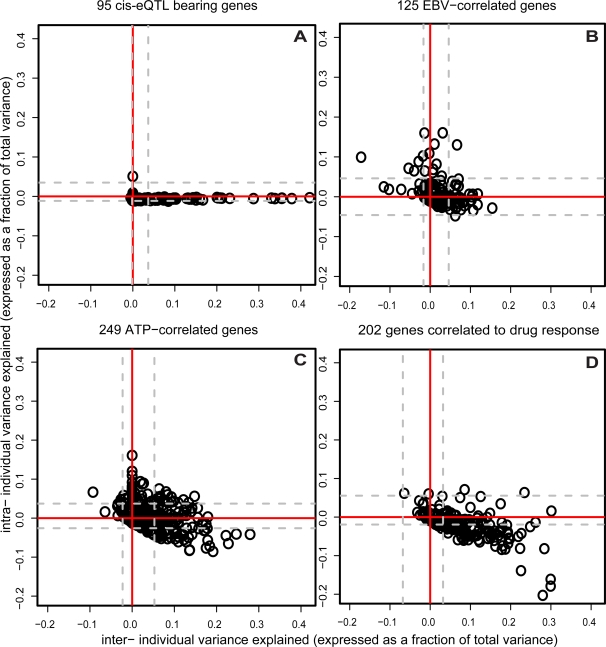
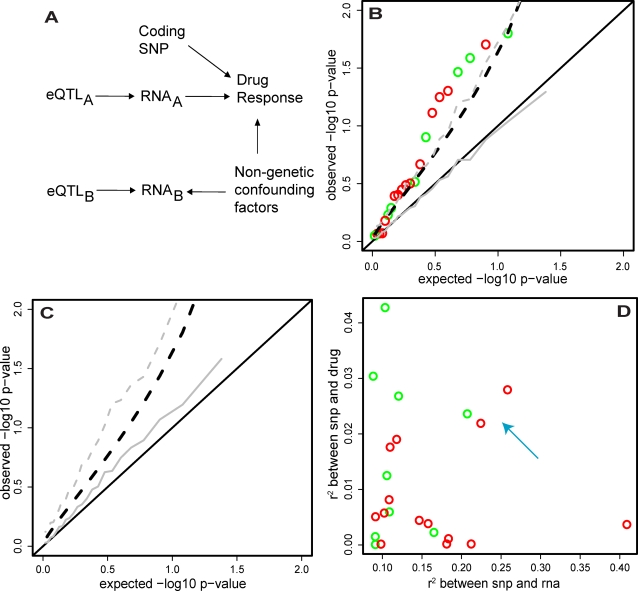
Lymphoblastoid cell lines (LCLs), originally collected as renewable sources of DNA, are now being used as a model system to study genotype-phenotype relationships in human cells, including searches for QTLs influencing levels of individual mRNAs and responses to drugs and radiation. In the course of attempting to map genes for drug response using 269 LCLs from the International HapMap Project, we evaluated the extent to which biological noise and non-genetic confounders contribute to trait variability in LCLs. While drug responses could be technically well measured on a given day, we observed significant day-to-day variability and substantial correlation to non-genetic confounders, such as baseline growth rates and metabolic state in culture. After correcting for these confounders, we were unable to detect any QTLs with genome-wide significance for drug response. A much higher proportion of variance in mRNA levels may be attributed to non-genetic factors (intra-individual variance--i.e., biological noise, levels of the EBV virus used to transform the cells, ATP levels) than to detectable eQTLs. Finally, in an attempt to improve power, we focused analysis on those genes that had both detectable eQTLs and correlation to drug response; we were unable to detect evidence that eQTL SNPs are convincingly associated with drug response in the model. While LCLs are a promising model for pharmacogenetic experiments, biological noise and in vitro artifacts may reduce power and have the potential to create spurious association due to confounding.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
-
- McCarthy MI, Abecasis GR, Cardon LR, Goldstein DB, Little J, et al. Genome-wide association studies for complex traits: consensus, uncertainty and challenges. Nat Rev Genet. 2008;9:356–369. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
