

The glucagon receptor is required for the adaptive metabolic response to fasting
- PMID: 19046568
- PMCID: PMC2593715
- DOI: 10.1016/j.cmet.2008.09.008
The glucagon receptor is required for the adaptive metabolic response to fasting
Abstract
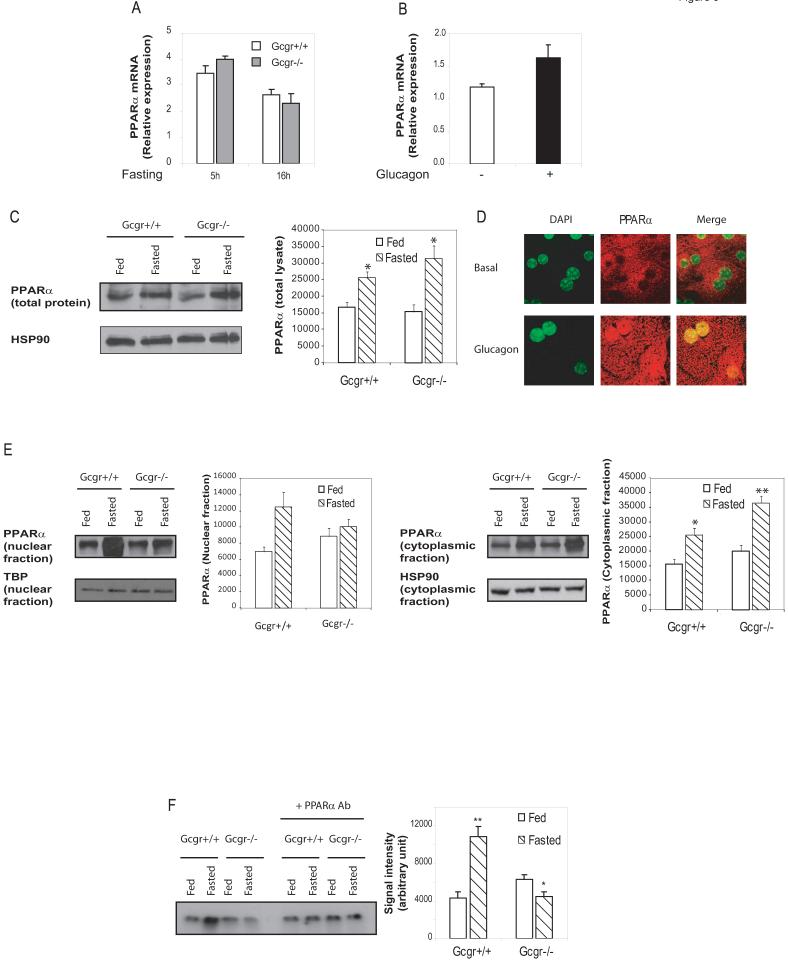
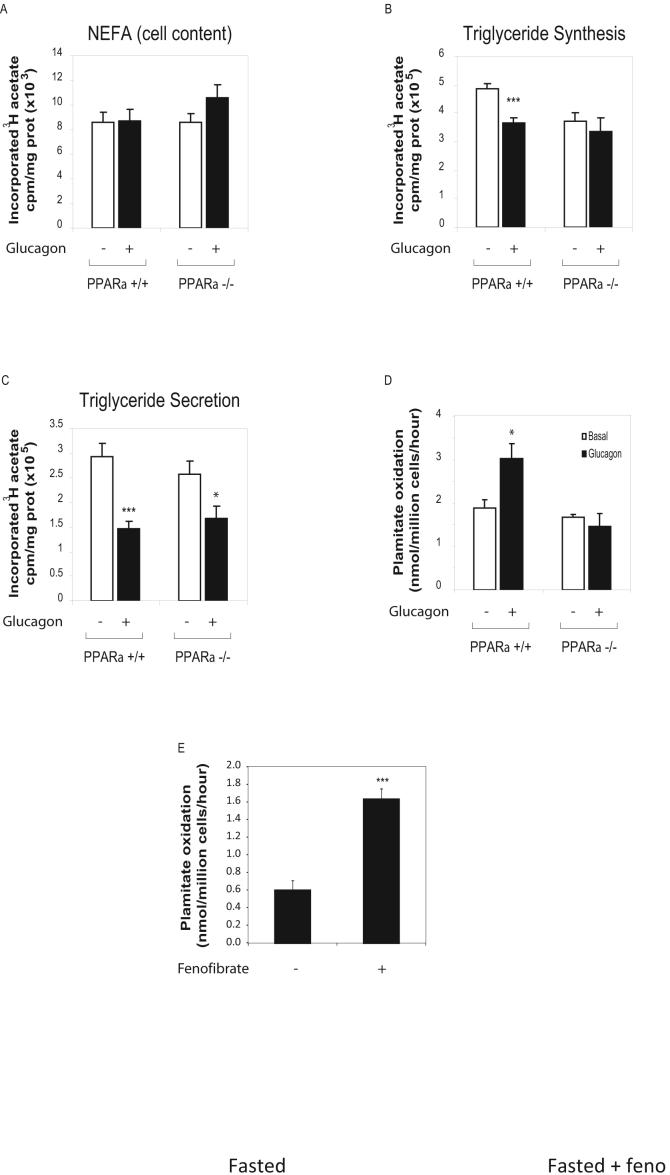
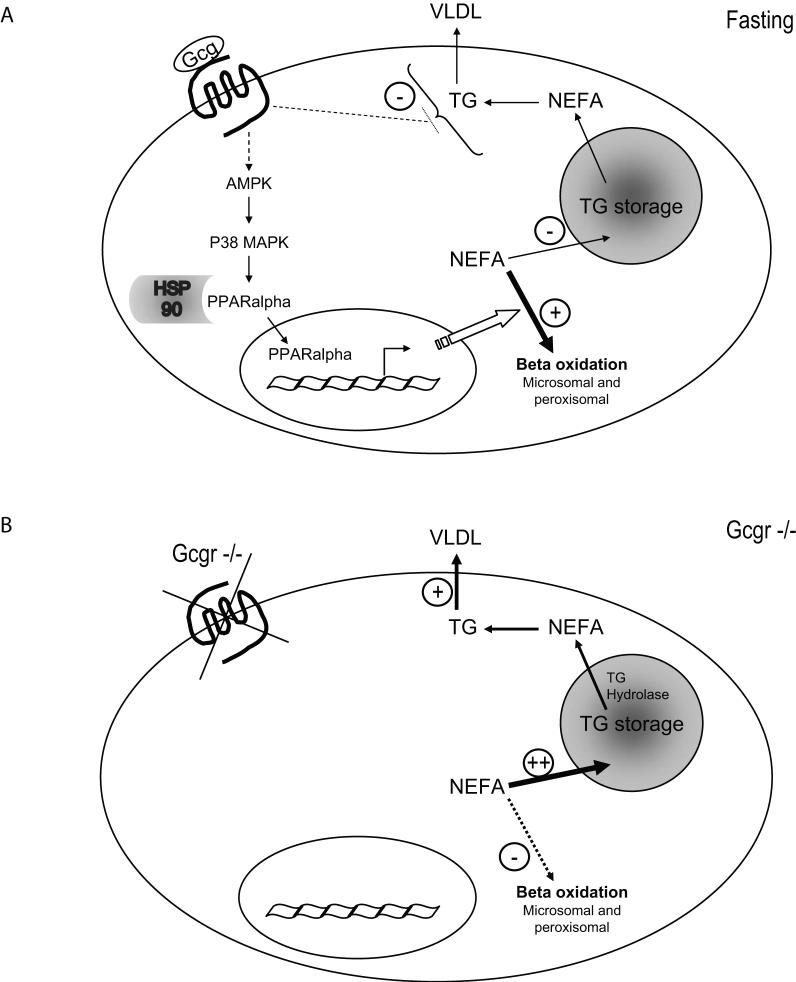
Glucagon receptor (Gcgr) signaling maintains hepatic glucose production during the fasting state; however, the importance of the Gcgr for lipid metabolism is unclear. We show here that fasted Gcgr-/- mice exhibit a significant increase in hepatic triglyceride secretion and fasting increases fatty acid oxidation (FAO) in wild-type (WT) but not in Gcgr-/- mice. Moreover fasting upregulated the expression of FAO-related hepatic mRNA transcripts in Gcgr+/+ but not in Gcgr-/- mice. Exogenous glucagon administration reduced plasma triglycerides in WT mice, inhibited TG synthesis and secretion, and stimulated FA beta oxidation in Gcgr+/+ hepatocytes. The actions of glucagon on TG synthesis and FAO were abolished in PPARalpha-/- hepatocytes. These findings demonstrate that the Gcgr receptor is required for control of lipid metabolism during the adaptive metabolic response to fasting.
Figures





References
-
- Bobe G, Ametaj BN, Young JW, Anderson LL, Beitz DC. Exogenous glucagon effects on health and reproductive performance of lactating dairy cows with mild fatty liver. Anim Reprod Sci. 2006 - PubMed
-
- Bobe G, Ametaj BN, Young JW, Beitz DC. Effects of exogenous glucagon on lipids in lipoproteins and liver of lactating dairy cows. J Dairy Sci. 2003a;86:2895–2903. - PubMed
-
- Bobe G, Ametaj BN, Young JW, Beitz DC. Potential treatment of fatty liver with 14-day subcutaneous injections of glucagon. J Dairy Sci. 2003b;86:3138–3147. - PubMed
-
- Cao W, Collins QF, Becker TC, Robidoux J, Lupo EG, Jr., Xiong Y, Daniel KW, Floering L, Collins S. p38 Mitogen-activated protein kinase plays a stimulatory role in hepatic gluconeogenesis. J Biol Chem. 2005;280:42731–42737. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
