

HDAC2 blockade by nitric oxide and histone deacetylase inhibitors reveals a common target in Duchenne muscular dystrophy treatment
- PMID: 19047631
- PMCID: PMC2614736
- DOI: 10.1073/pnas.0805514105
HDAC2 blockade by nitric oxide and histone deacetylase inhibitors reveals a common target in Duchenne muscular dystrophy treatment
Erratum in
- Proc Natl Acad Sci U S A. 2009 Feb 3;106(5):1679. Steinkulher, Christian [corrected to Steinkuhler, Christian]
Abstract
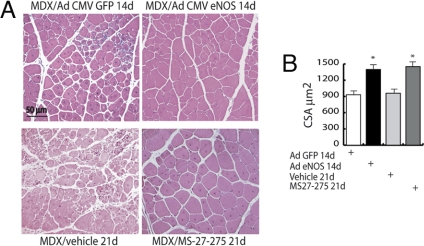
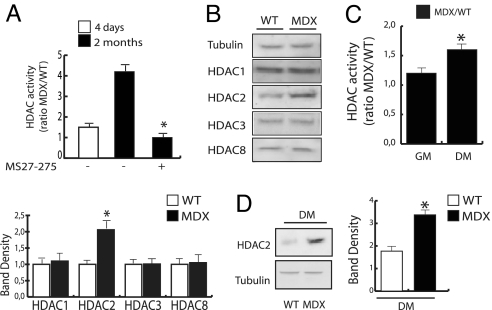
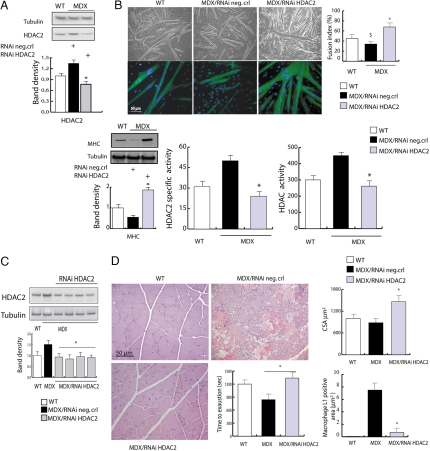
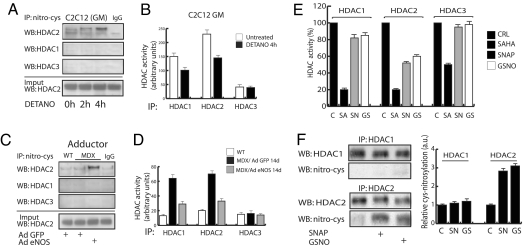
The overlapping histological and biochemical features underlying the beneficial effect of deacetylase inhibitors and NO donors in dystrophic muscles suggest an unanticipated molecular link among dystrophin, NO signaling, and the histone deacetylases (HDACs). Higher global deacetylase activity and selective increased expression of the class I histone deacetylase HDAC2 were detected in muscles of dystrophin-deficient MDX mice. In vitro and in vivo siRNA-mediated down-regulation of HDAC2 in dystrophic muscles was sufficient to replicate the morphological and functional benefits observed with deacetylase inhibitors and NO donors. We found that restoration of NO signaling in vivo, by adenoviral-mediated expression of a constitutively active endothelial NOS mutant in MDX muscles, and in vitro, by exposing MDX-derived satellite cells to NO donors, resulted in HDAC2 blockade by cysteine S-nitrosylation. These data reveal a special contribution of HDAC2 in the pathogenesis of Duchenne muscular dystrophy and indicate that HDAC2 inhibition by NO-dependent S-nitrosylation is important for the therapeutic response to NO donors in MDX mice. They also define a common target for independent pharmacological interventions in the treatment of Duchenne muscular dystrophy.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Minetti GC, et al. Functional and morphological recovery of dystrophic muscles in mice treated with deacetylase inhibitors. Nat Med. 2006;12:1147–1150. - PubMed
-
- Iezzi S, et al. Deacetylase inhibitors increase muscle cell size by promoting myoblast recruitment and fusion through induction of follistatin. Dev Cell. 2004;6:673–684. - PubMed
-
- Bogdanovich S, et al. Functional improvement of dystrophic muscle by myostatin blockade. Nature. 2002;420:418–421. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
