

Genetic variants of Nogo-66 receptor with possible association to schizophrenia block myelin inhibition of axon growth
- PMID: 19052207
- PMCID: PMC2892845
- DOI: 10.1523/JNEUROSCI.3828-08.2008
Genetic variants of Nogo-66 receptor with possible association to schizophrenia block myelin inhibition of axon growth
Abstract
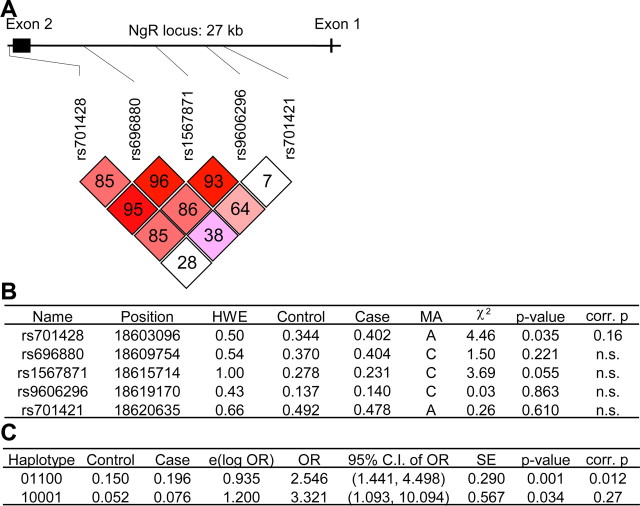
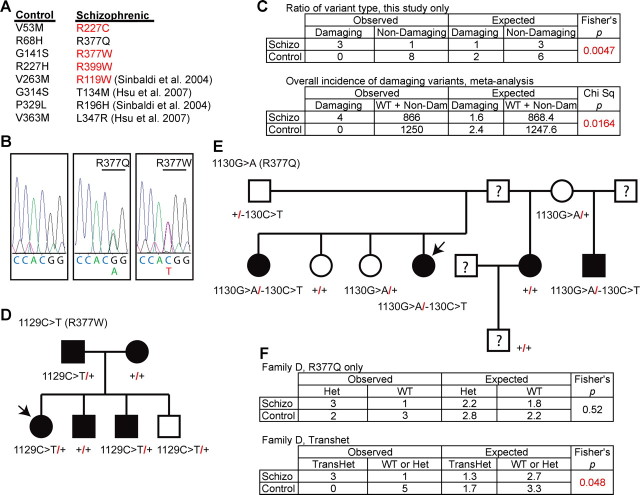
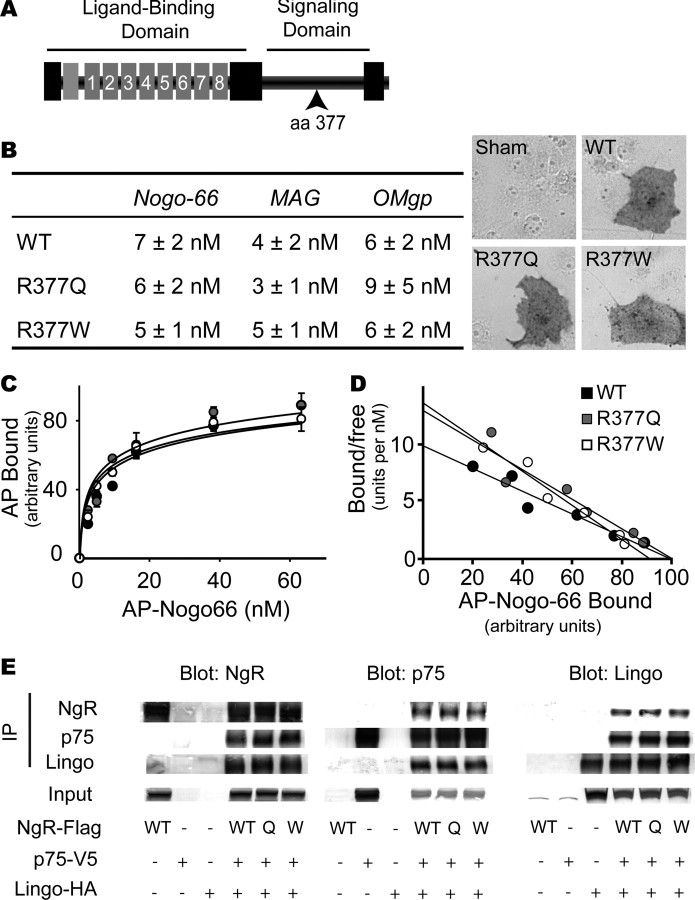
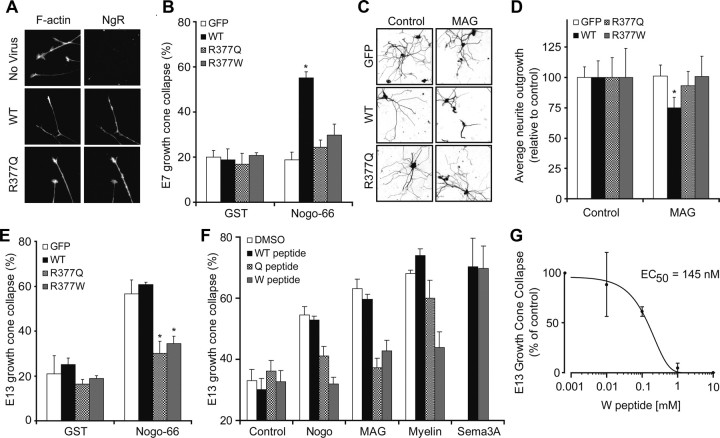
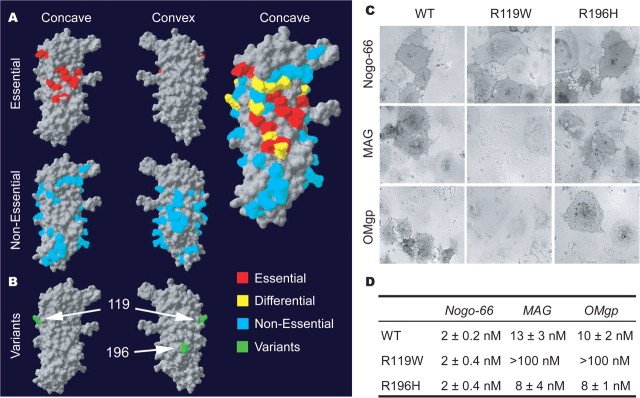
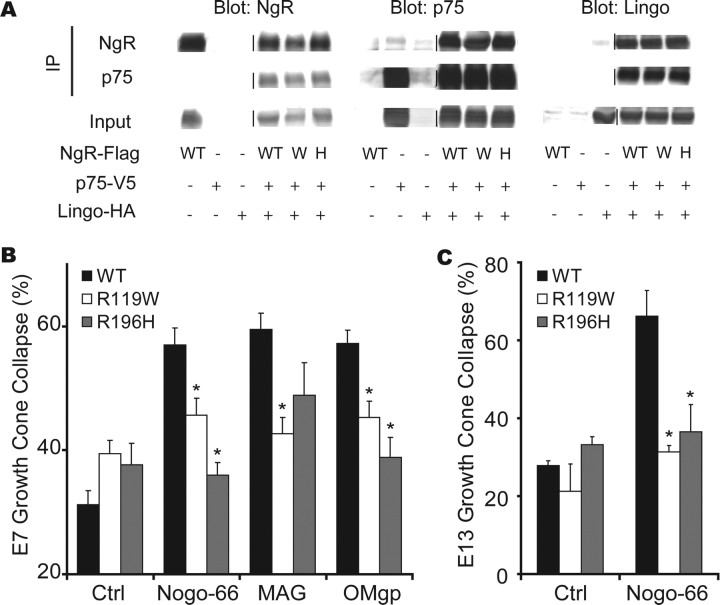
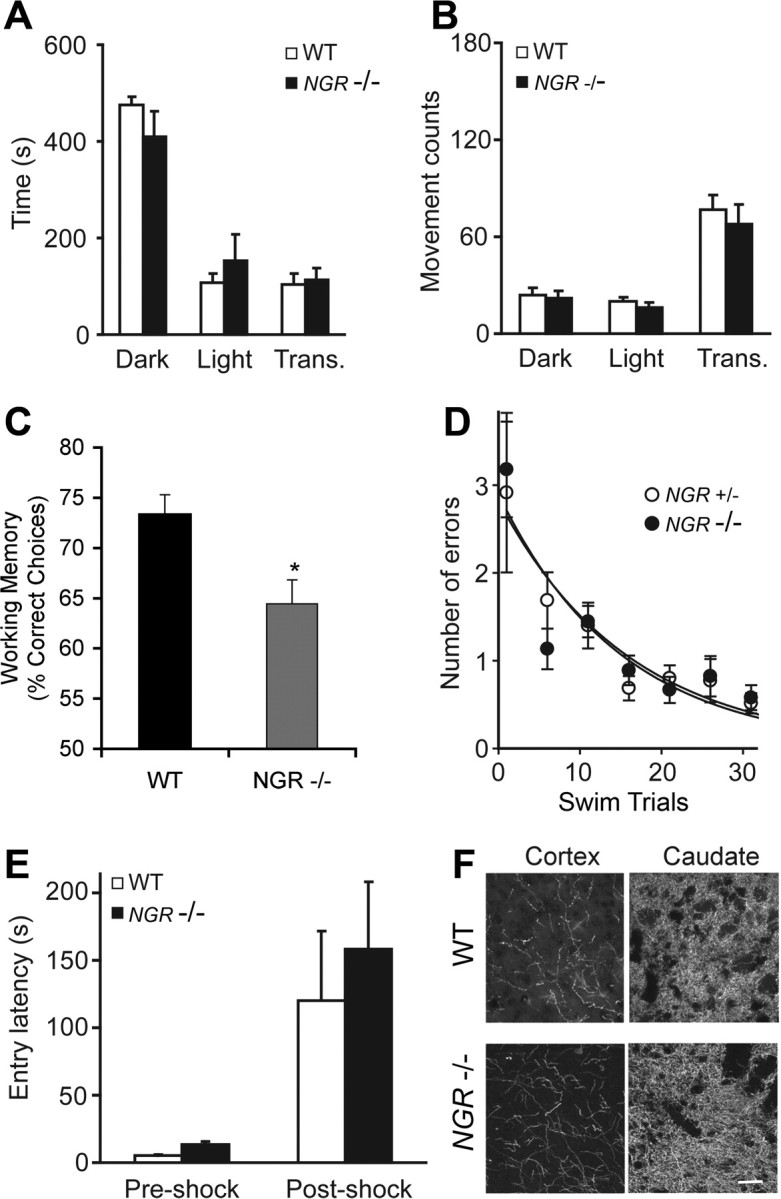
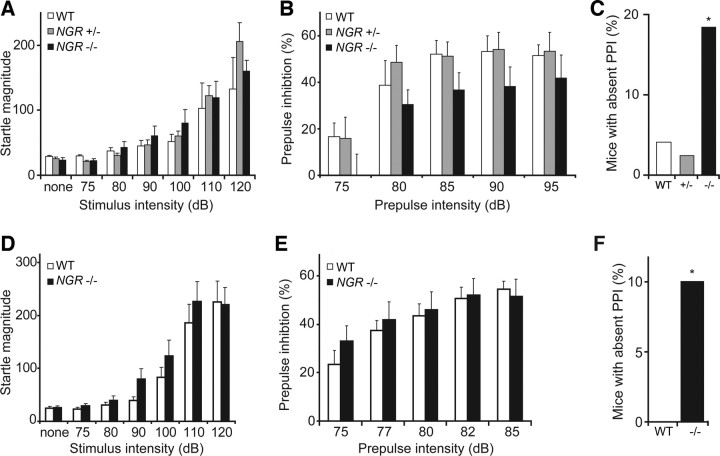
In schizophrenia, genetic predisposition has been linked to chromosome 22q11 and myelin-specific genes are misexpressed in schizophrenia. Nogo-66 receptor 1 (NGR or RTN4R) has been considered to be a 22q11 candidate gene for schizophrenia susceptibility because it encodes an axonal protein that mediates myelin inhibition of axonal sprouting. Confirming previous studies, we found that variation at the NGR locus is associated with schizophrenia in a Caucasian case-control analysis, and this association is not attributed to population stratification. Within a limited set of schizophrenia-derived DNA samples, we identified several rare NGR nonconservative coding sequence variants. Neuronal cultures demonstrate that four different schizophrenia-derived NgR1 variants fail to transduce myelin signals into axon inhibition, and function as dominant negatives to disrupt endogenous NgR1. This provides the first evidence that certain disease-derived human NgR1 variants are dysfunctional proteins in vitro. Mice lacking NgR1 protein exhibit reduced working memory function, consistent with a potential endophenotype of schizophrenia. For a restricted subset of individuals diagnosed with schizophrenia, the expression of dysfunctional NGR variants may contribute to increased disease risk.
Figures
Comment in
-
Converging evidence for the Nogo-66 receptor gene in schizophrenia.J Neurosci. 2009 Apr 22;29(16):5045-7. doi: 10.1523/JNEUROSCI.0477-09.2009. J Neurosci. 2009. PMID: 19386899 Free PMC article. Review. No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases