

Computational and experimental approaches to chart the Escherichia coli cell-envelope-associated proteome and interactome
- PMID: 19054114
- PMCID: PMC2704936
- DOI: 10.1111/j.1574-6976.2008.00141.x
Computational and experimental approaches to chart the Escherichia coli cell-envelope-associated proteome and interactome
Abstract
The bacterial cell-envelope consists of a complex arrangement of lipids, proteins and carbohydrates that serves as the interface between a microorganism and its environment or, with pathogens, a human host. Escherichia coli has long been investigated as a leading model system to elucidate the fundamental mechanisms underlying microbial cell-envelope biology. This includes extensive descriptions of the molecular identities, biochemical activities and evolutionary trajectories of integral transmembrane proteins, many of which play critical roles in infectious disease and antibiotic resistance. Strikingly, however, only half of the c. 1200 putative cell-envelope-related proteins of E. coli currently have experimentally attributed functions, indicating an opportunity for discovery. In this review, we summarize the state of the art of computational and proteomic approaches for determining the components of the E. coli cell-envelope proteome, as well as exploring the physical and functional interactions that underlie its biogenesis and functionality. We also provide a comprehensive comparative benchmarking analysis on the performance of different bioinformatic and proteomic methods commonly used to determine the subcellular localization of bacterial proteins.
Figures
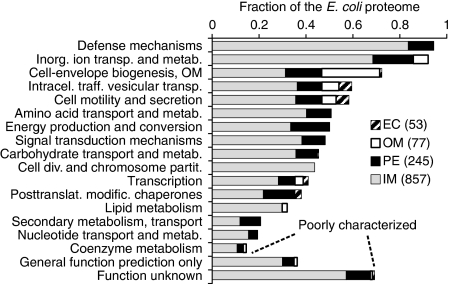
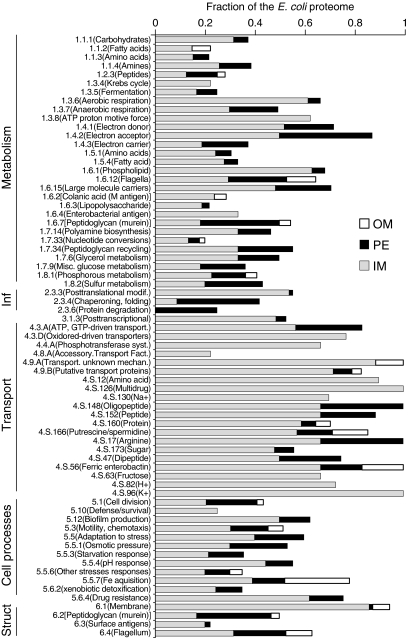


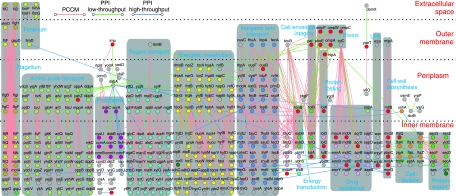
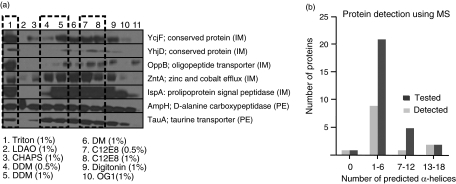
References
-
- Aggarwal K, Choe LH, Lee KH. Quantitative analysis of protein expression using amine-specific isobaric tags in Escherichia coli cells expressing rhsA elements. Proteomics. 2005;5:2297–2308. - PubMed
-
- Andrade MA, Brown NP, Leroy C, et al. Automated genome sequence analysis and annotation. Bioinformatics. 1999;15:391–412. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
