

RNA cytosine methylation analysis by bisulfite sequencing
- PMID: 19059995
- PMCID: PMC2632927
- DOI: 10.1093/nar/gkn954
RNA cytosine methylation analysis by bisulfite sequencing
Abstract
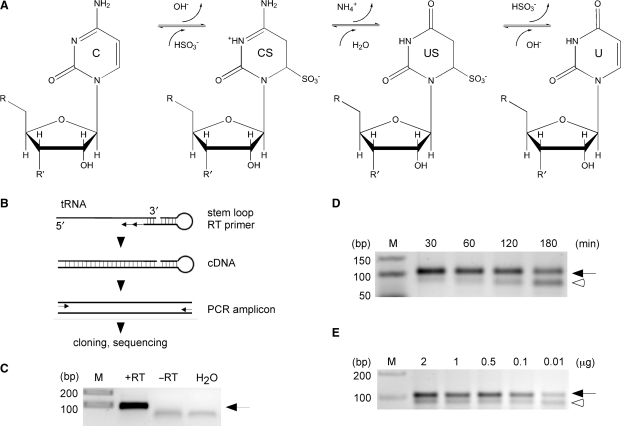
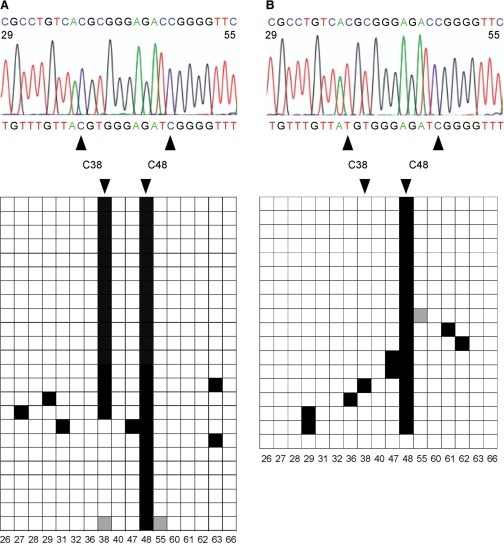
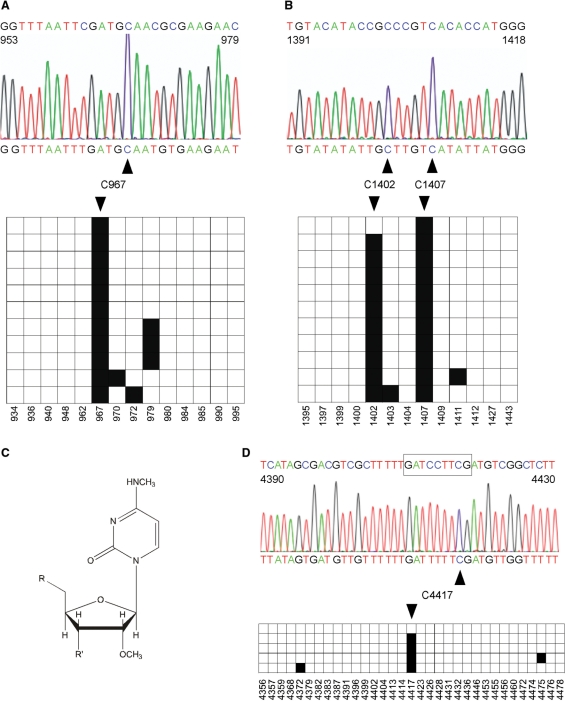
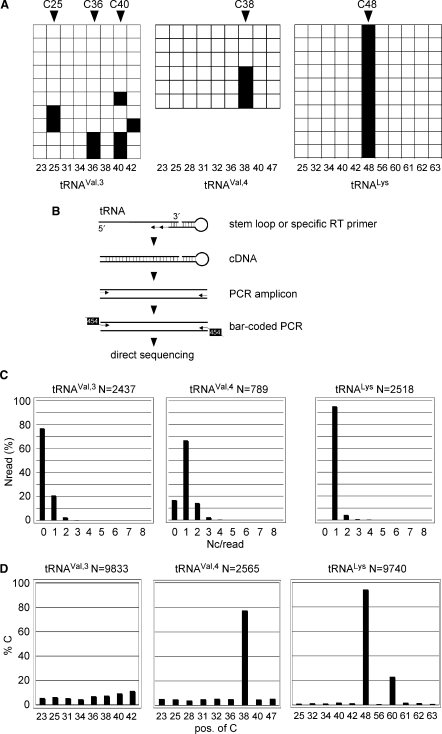
Covalent modifications of nucleic acids play an important role in regulating their functions. Among these modifications, (cytosine-5) DNA methylation is best known for its role in the epigenetic regulation of gene expression. Post-transcriptional RNA modification is a characteristic feature of noncoding RNAs, and has been described for rRNAs, tRNAs and miRNAs. (Cytosine-5) RNA methylation has been detected in stable and long-lived RNA molecules, but its function is still unclear, mainly due to technical limitations. In order to facilitate the analysis of RNA methylation patterns we have established a protocol for the chemical deamination of cytosines in RNA, followed by PCR-based amplification of cDNA and DNA sequencing. Using tRNAs and rRNAs as examples we show that cytosine methylation can be reproducibly and quantitatively detected by bisulfite sequencing. The combination of this method with deep sequencing allowed the analysis of a large number of RNA molecules. These results establish a versatile method for the identification and characterization of RNA methylation patterns, which will be useful for defining the biological function of RNA methylation.
Figures

References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
