

A genetic interaction map of RNA-processing factors reveals links between Sem1/Dss1-containing complexes and mRNA export and splicing
- PMID: 19061648
- PMCID: PMC2644724
- DOI: 10.1016/j.molcel.2008.11.012
A genetic interaction map of RNA-processing factors reveals links between Sem1/Dss1-containing complexes and mRNA export and splicing
Abstract
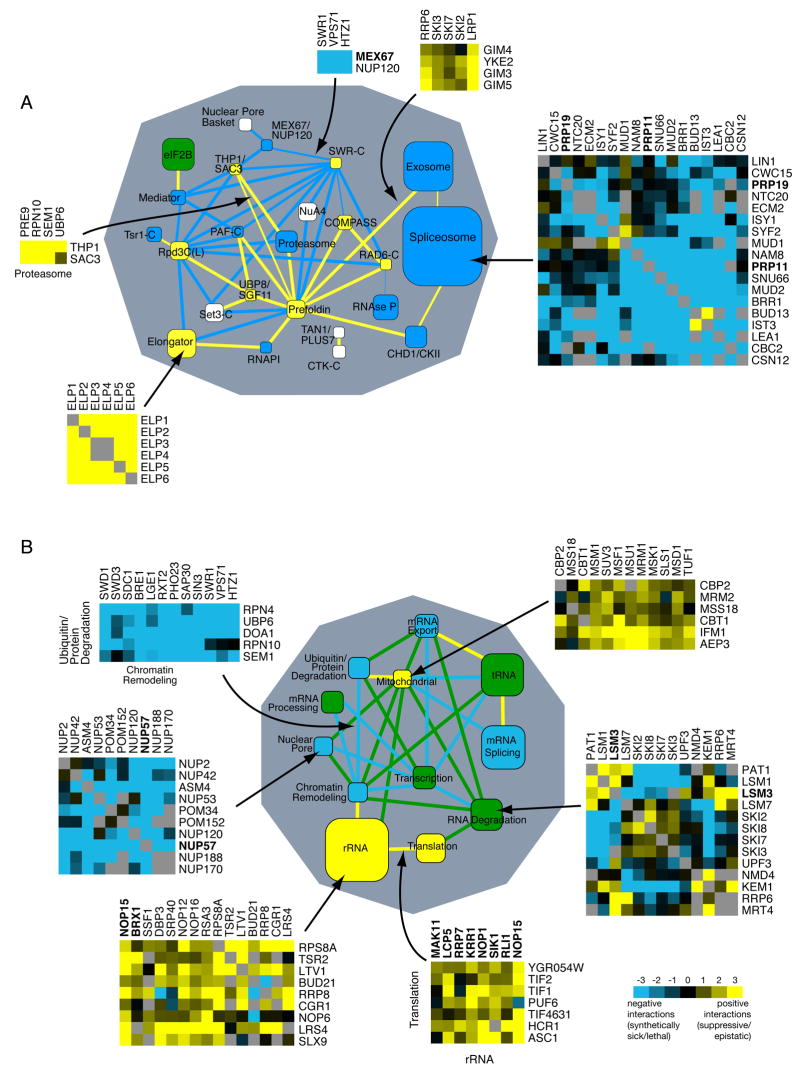
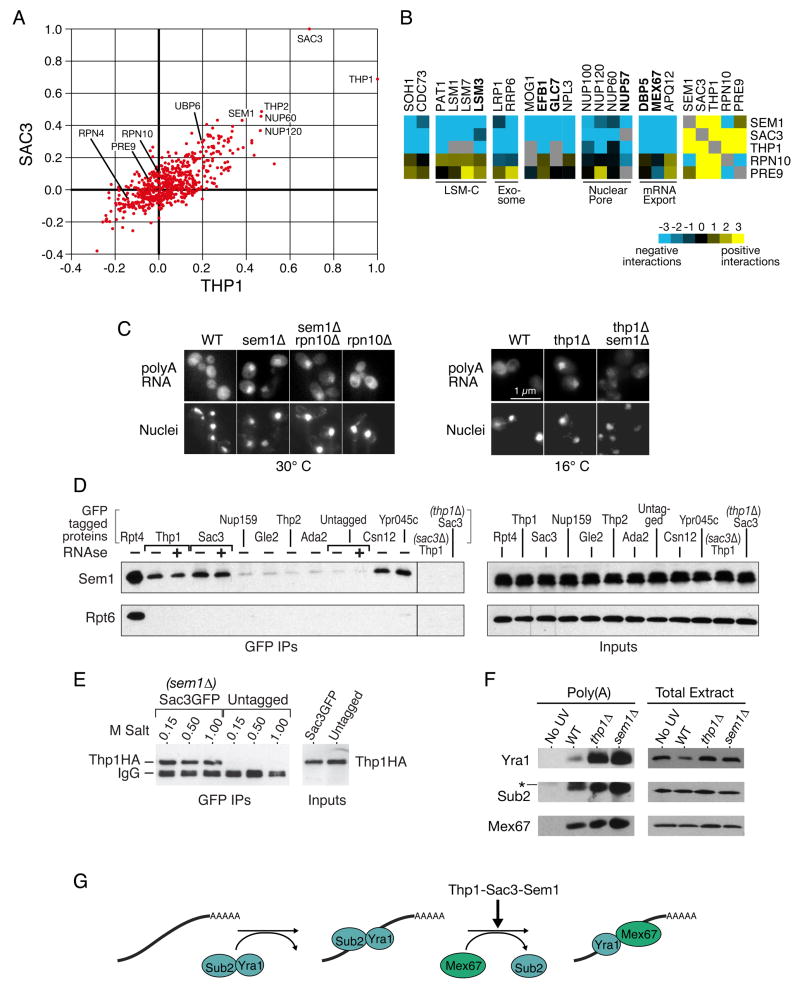
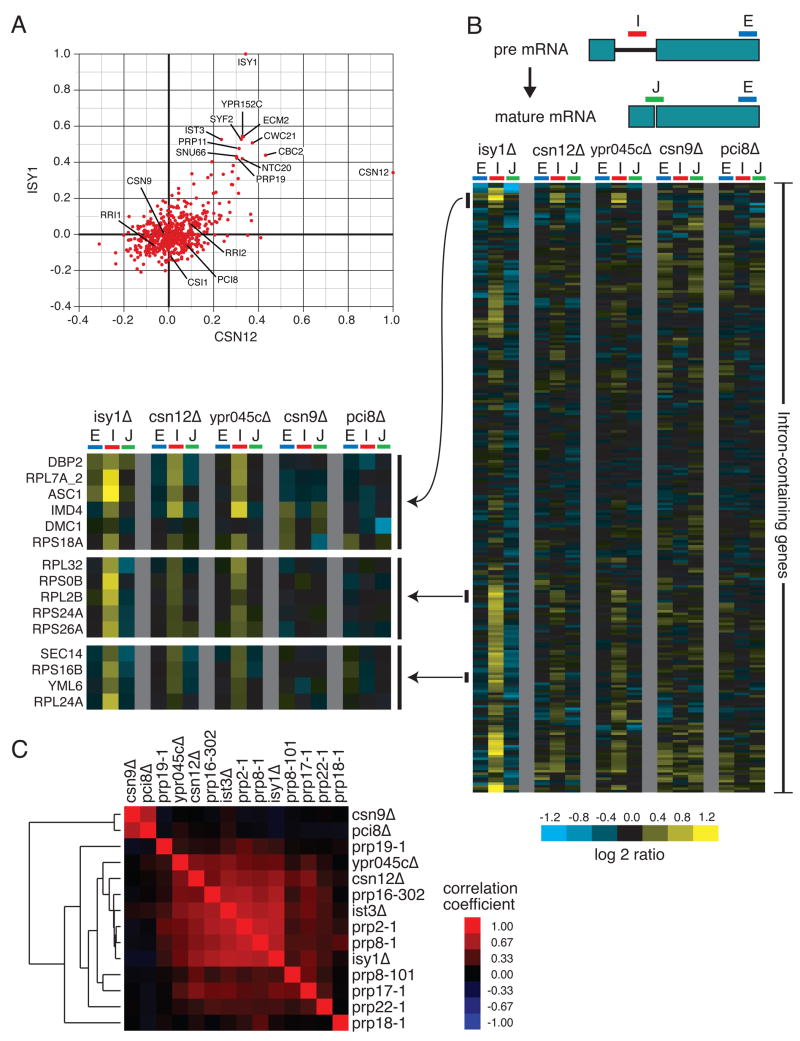
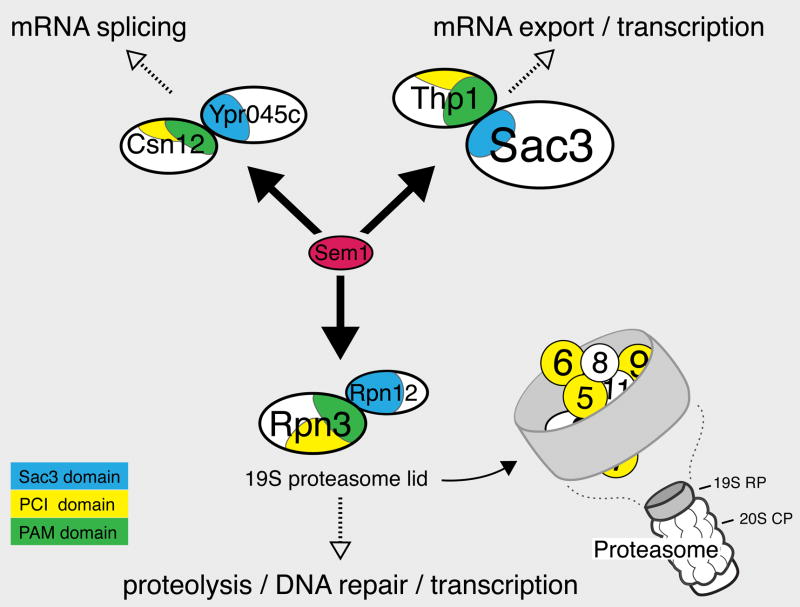
We used a quantitative, high-density genetic interaction map, or E-MAP (Epistatic MiniArray Profile), to interrogate the relationships within and between RNA-processing pathways. Due to their complexity and the essential roles of many of the components, these pathways have been difficult to functionally dissect. Here, we report the results for 107,155 individual interactions involving 552 mutations, 166 of which are hypomorphic alleles of essential genes. Our data enabled the discovery of links between components of the mRNA export and splicing machineries and Sem1/Dss1, a component of the 19S proteasome. In particular, we demonstrate that Sem1 has a proteasome-independent role in mRNA export as a functional component of the Sac3-Thp1 complex. Sem1 also interacts with Csn12, a component of the COP9 signalosome. Finally, we show that Csn12 plays a role in pre-mRNA splicing, which is independent of other signalosome components. Thus, Sem1 is involved in three separate and functionally distinct complexes.
Figures

References
-
- Baillat D, Hakimi MA, Naar AM, Shilatifard A, Cooch N, Shiekhattar R. Integrator, a multiprotein mediator of small nuclear RNA processing, associates with the C-terminal repeat of RNA polymerase II. Cell. 2005;123:265–276. - PubMed
-
- Becker RA, Chambers JM, Wilks AR. The new S language: a programming environment for data analysis and graphics. Pacific Grove, Calif.: Wadsworth & Brooks/Cole Advanced Books & Software; 1988.
-
- Chan SP, Kao DI, Tsai WY, Cheng SC. The Prp19p-associated complex in spliceosome activation. Science. 2003;302:279–282. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
