

Akt determines replicative senescence and oxidative or oncogenic premature senescence and sensitizes cells to oxidative apoptosis
- PMID: 19061837
- PMCID: PMC3038665
- DOI: 10.1016/j.ccr.2008.11.003
Akt determines replicative senescence and oxidative or oncogenic premature senescence and sensitizes cells to oxidative apoptosis
Abstract
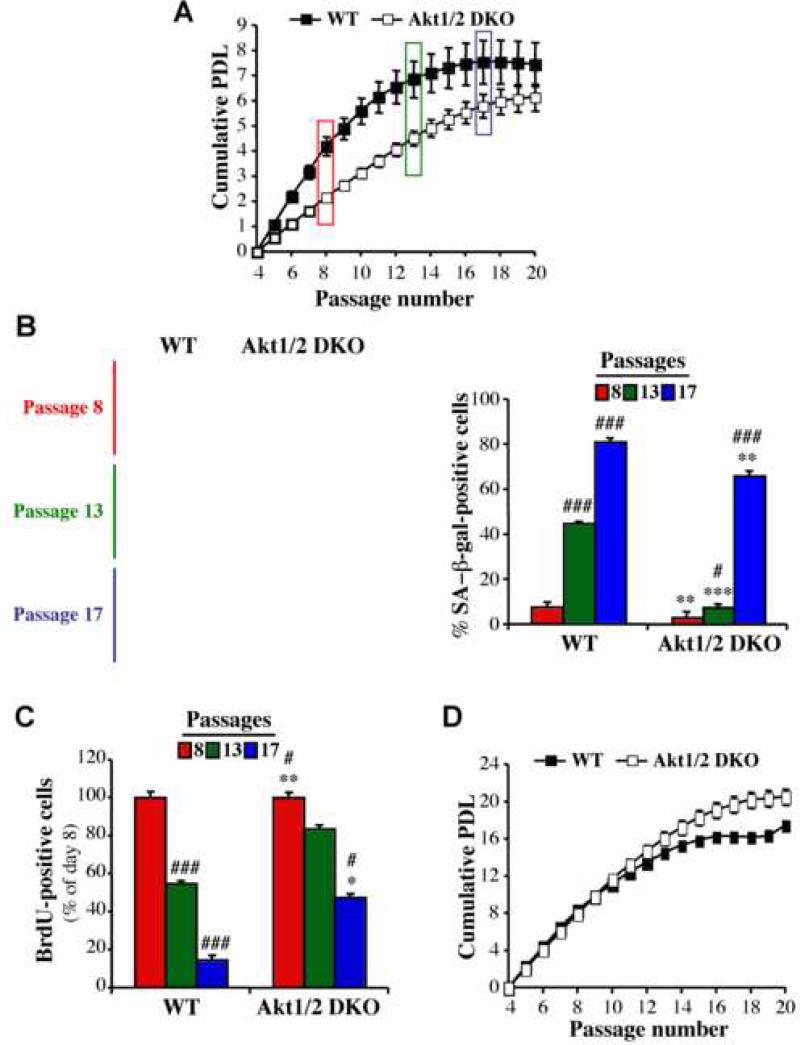
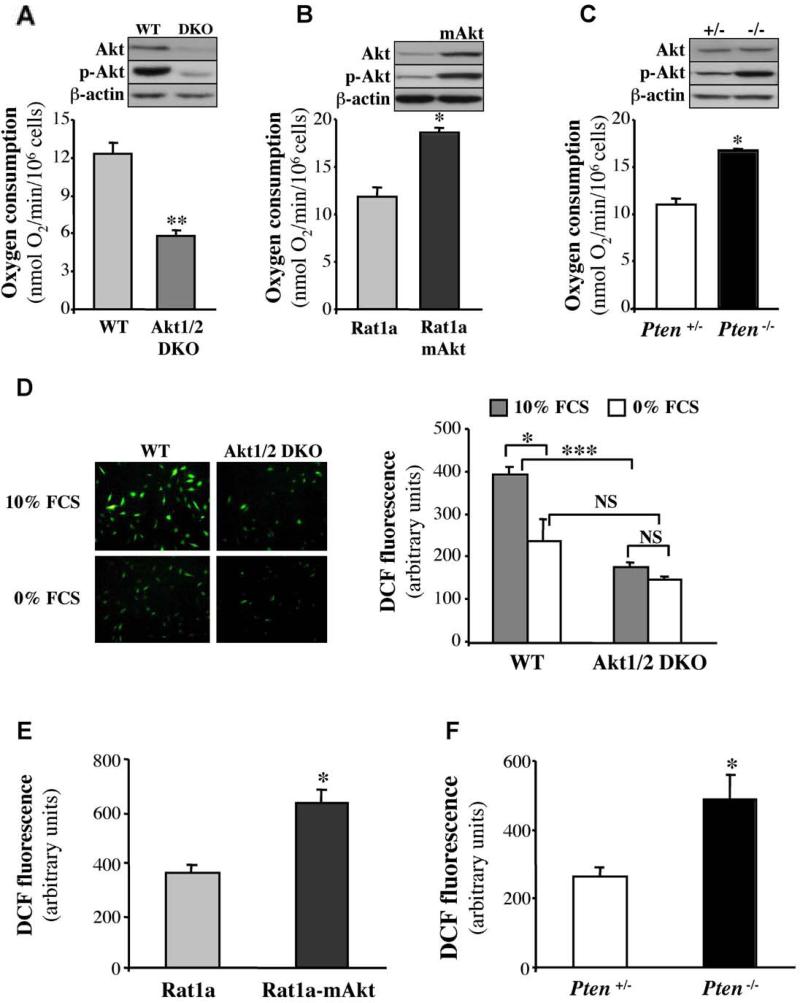
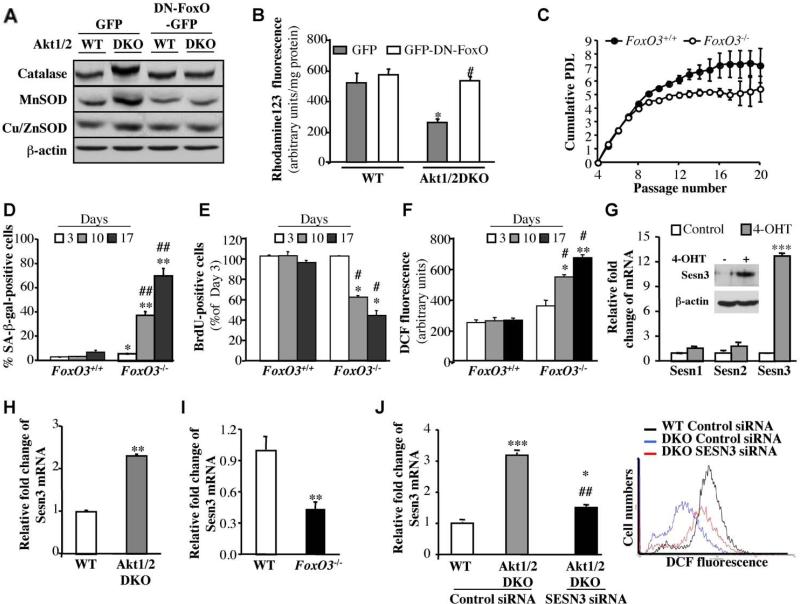
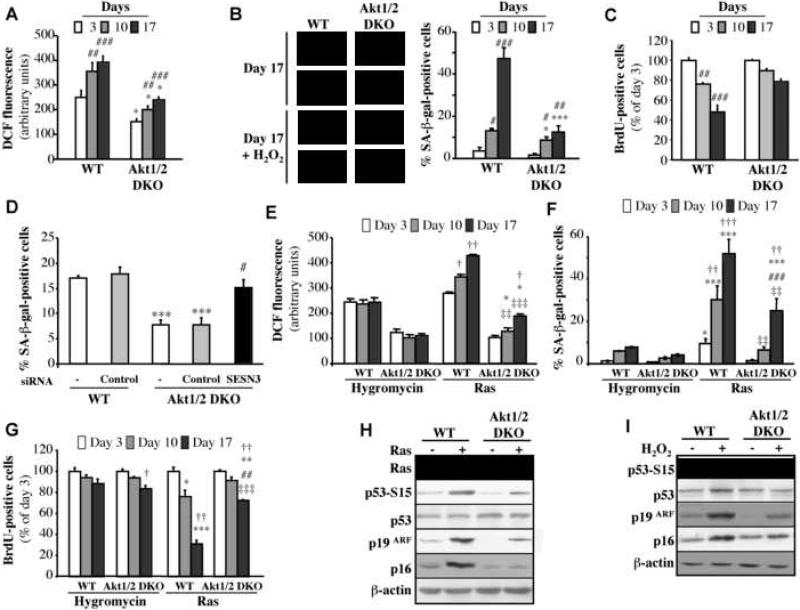
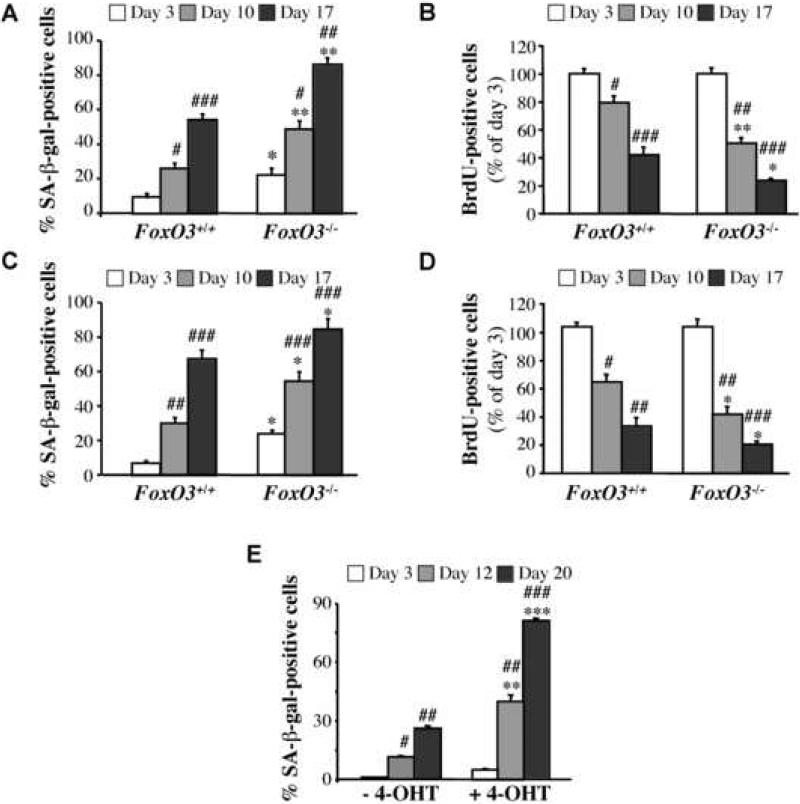
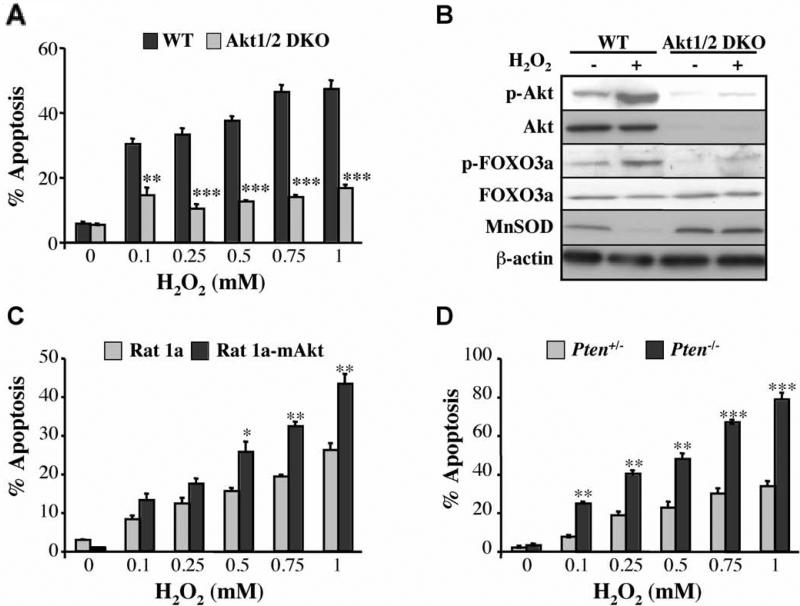
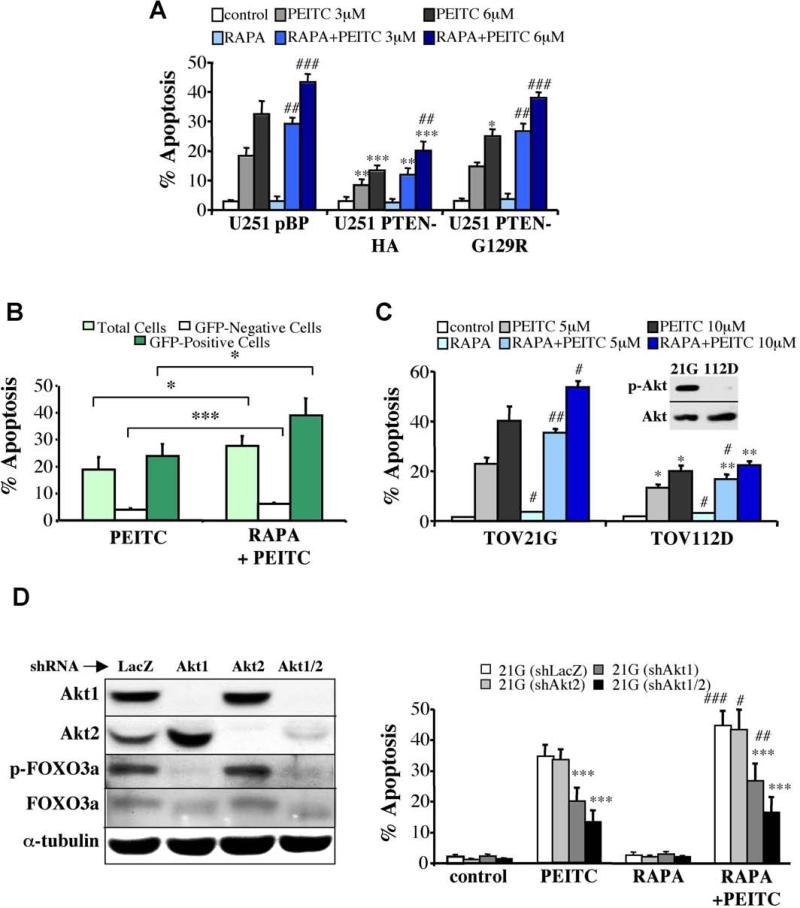
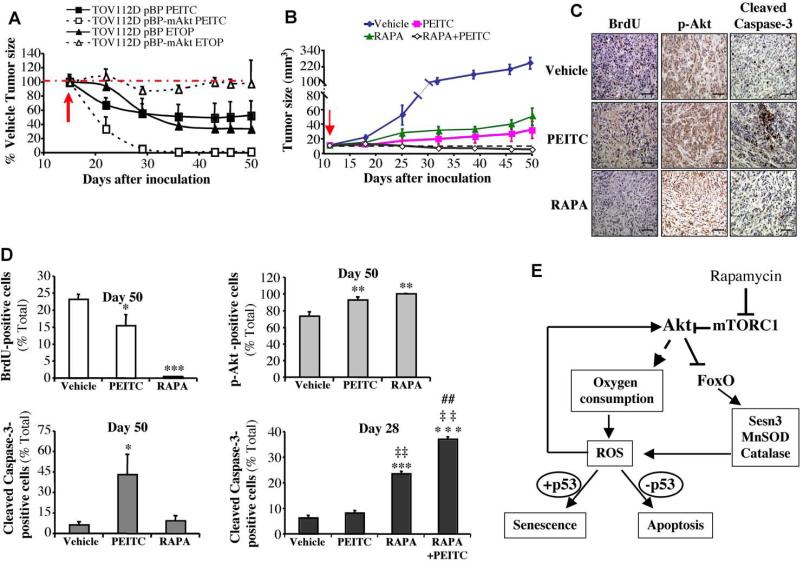
Akt deficiency causes resistance to replicative senescence, to oxidative stress- and oncogenic Ras-induced premature senescence, and to reactive oxygen species (ROS)-mediated apoptosis. Akt activation induces premature senescence and sensitizes cells to ROS-mediated apoptosis by increasing intracellular ROS through increased oxygen consumption and by inhibiting the expression of ROS scavengers downstream of FoxO, particularly sestrin 3. This uncovers an Achilles' heel of Akt, since in contrast to its ability to inhibit apoptosis induced by multiple apoptotic stimuli, Akt could not inhibit ROS-mediated apoptosis. Furthermore, treatment with rapamycin that led to further Akt activation and resistance to etoposide hypersensitized cancer cells to ROS-mediated apoptosis. Given that rapamycin alone is mainly cytostatic, this constitutes a strategy for cancer therapy that selectively eradicates cancer cells via Akt activation.
Figures
Comment in
-
AKT and oxidative stress team up to kill cancer cells.Cancer Cell. 2008 Dec 9;14(6):427-9. doi: 10.1016/j.ccr.2008.11.006. Cancer Cell. 2008. PMID: 19061832
References
-
- Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. 2005;120:483–495. - PubMed
-
- Bhaskar PT, Hay N. The Two TORCs and Akt. Dev Cell. 2007;12:487–502. - PubMed
-
- Braig M, Lee S, Loddenkemper C, Rudolph C, Peters AH, Schlegelberger B, Stein H, Dorken B, Jenuwein T, Schmitt CA. Oncogene-induced senescence as an initial barrier in lymphoma development. Nature. 2005;436:660–665. - PubMed
-
- Budanov AV, Sablina AA, Feinstein E, Koonin EV, Chumakov PM. Regeneration of peroxiredoxins by p53-regulated sestrins, homologs of bacterial AhpD. Science. 2004;304:596–600. - PubMed
-
- Budanov AV, Shoshani T, Faerman A, Zelin E, Kamer I, Kalinski H, Gorodin S, Fishman A, Chajut A, Einat P, et al. Identification of a novel stress-responsive gene Hi95 involved in regulation of cell viability. Oncogene. 2002;21:6017–6031. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
