

A missense mutation in SLC33A1, which encodes the acetyl-CoA transporter, causes autosomal-dominant spastic paraplegia (SPG42)
- PMID: 19061983
- PMCID: PMC2668077
- DOI: 10.1016/j.ajhg.2008.11.003
A missense mutation in SLC33A1, which encodes the acetyl-CoA transporter, causes autosomal-dominant spastic paraplegia (SPG42)
Abstract
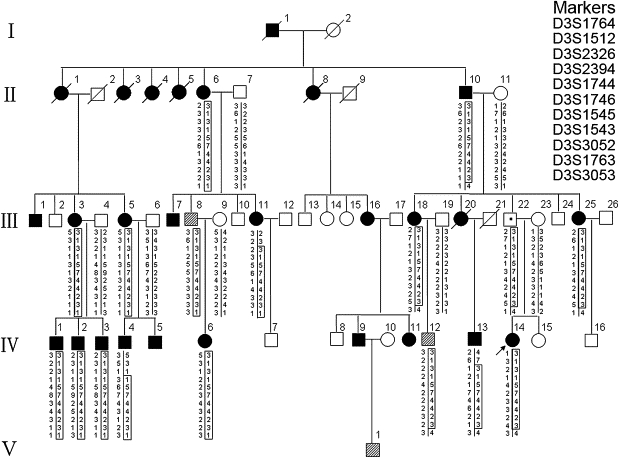
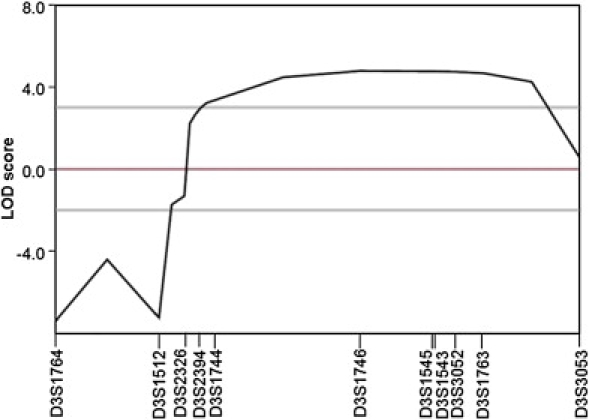
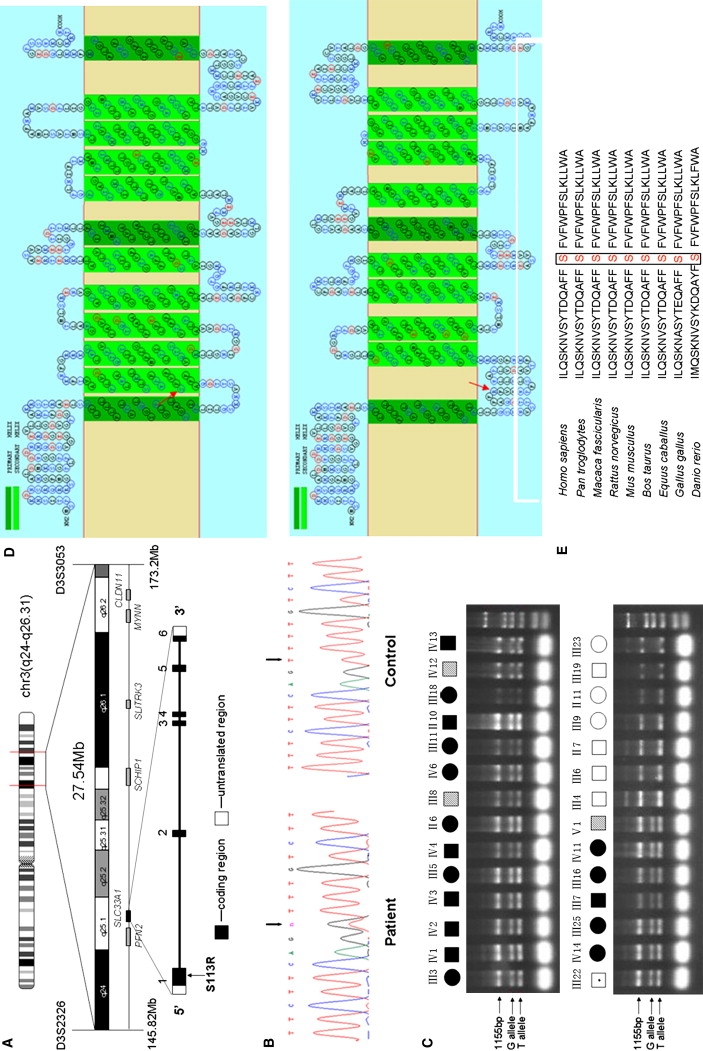
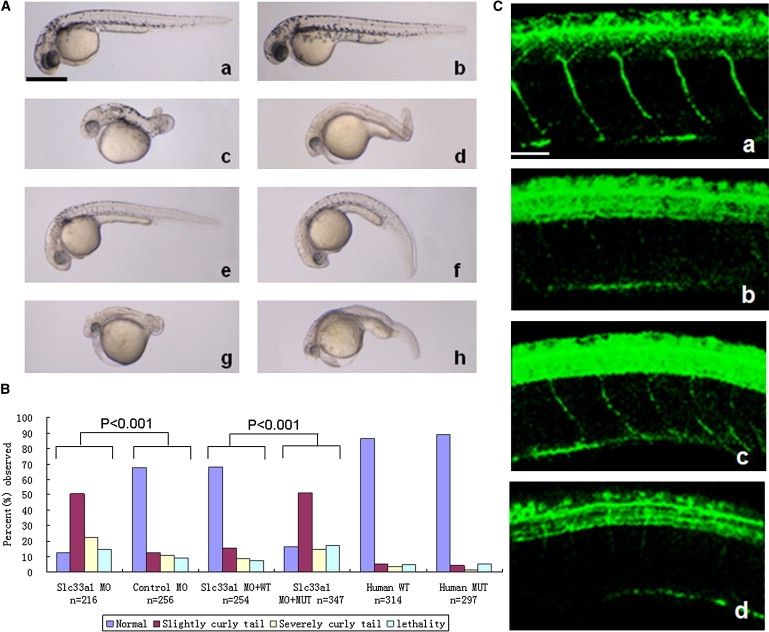
Hereditary spastic paraplegias (HSPs), characterized by progressive and bilateral spasticity of the legs, are usually caused by developmental failure or degeneration of motor axons in the corticospinal tract. There are considerable interfamilial and intrafamilial variations in age at onset and severity of spasticity. Genetic studies also showed that there are dozens of genetic loci, on multiple chromosomes, that are responsible for HSPs. Through linkage study of a pedigree of HSP with autosomal-dominant inheritance, we mapped the causative gene to 3q24-q26. Screening of candidate genes revealed that the HSP is caused by a missense mutation in the gene for acetyl-CoA transporter (SLC33A1). It is predicted that the missense mutation, causing the change of the highly conserved serine to arginine at the codon 113 (p. S113R), disrupts the second transmembrane domain in the transporter and reverses the orientation of all of the descending domains. Knockdown of Slc33a1 in zebrafish caused a curve-shaped tail and defective axon outgrowth from the spinal cord. Although the wild-type human SLC33A1 was able to rescue the phenotype caused by Slc33a1 knockdown in zebrafish, the mutant SLC33A1 (p.S113R) was not, suggesting that S113R mutation renders SLC33A1 nonfunctional and one that wild-type allele is not sufficient for sustaining the outgrowth and maintenance of long motor axons in human heterozygotes. Thus, our study illustrated a critical role of acetyl-CoA transporter in motor-neuron development and function.
Figures
Similar articles
-
S113R mutation in SLC33A1 leads to neurodegeneration and augmented BMP signaling in a mouse model.Dis Model Mech. 2017 Jan 1;10(1):53-62. doi: 10.1242/dmm.026880. Epub 2016 Nov 24. Dis Model Mech. 2017. PMID: 27935820 Free PMC article.
-
A total of 220 patients with autosomal dominant spastic paraplegia do not display mutations in the SLC33A1 gene (SPG42).Eur J Hum Genet. 2010 Sep;18(9):1065-7. doi: 10.1038/ejhg.2010.68. Epub 2010 May 12. Eur J Hum Genet. 2010. PMID: 20461110 Free PMC article.
-
Identification and functional analysis of a SLC33A1: c.339T>G (p.Ser113Arg) variant in the original SPG42 family.Hum Mutat. 2015 Feb;36(2):240-9. doi: 10.1002/humu.22732. Hum Mutat. 2015. PMID: 25402622
-
Evidence of kinesin heavy chain (KIF5A) involvement in pure hereditary spastic paraplegia.Neurology. 2004 Sep 28;63(6):1108-10. doi: 10.1212/01.wnl.0000138731.60693.d2. Neurology. 2004. PMID: 15452312 Review.
-
Hereditary spastic paraplegia: advances in genetic research. Hereditary Spastic Paraplegia Working group.Neurology. 1996 Jun;46(6):1507-14. doi: 10.1212/wnl.46.6.1507. Neurology. 1996. PMID: 8649538 Review.
Cited by
-
Genetics of motor neuron disorders: new insights into pathogenic mechanisms.Nat Rev Genet. 2009 Nov;10(11):769-82. doi: 10.1038/nrg2680. Epub 2009 Oct 13. Nat Rev Genet. 2009. PMID: 19823194 Review.
-
Increased expression of SLC25A1/CIC causes an autistic-like phenotype with altered neuron morphology.Brain. 2022 Apr 18;145(2):500-516. doi: 10.1093/brain/awab295. Brain. 2022. PMID: 35203088 Free PMC article.
-
Deficient import of acetyl-CoA into the ER lumen causes neurodegeneration and propensity to infections, inflammation, and cancer.J Neurosci. 2014 May 14;34(20):6772-89. doi: 10.1523/JNEUROSCI.0077-14.2014. J Neurosci. 2014. PMID: 24828632 Free PMC article.
-
Machines like us scientists? : AI tools for mining the scientific literature in basic biomedical science.EMBO Rep. 2025 Aug;26(15):3709-3713. doi: 10.1038/s44319-025-00522-5. Epub 2025 Jul 10. EMBO Rep. 2025. PMID: 40640423 Free PMC article.
-
Genome-wide association study identifies novel single nucleotide polymorphisms having age-specific effect on prostate-specific antigen levels.Prostate. 2020 Dec;80(16):1405-1412. doi: 10.1002/pros.24070. Epub 2020 Sep 11. Prostate. 2020. PMID: 32914890 Free PMC article.
References
-
- Harding A.E. Classification of the hereditary ataxias and paraplegias. Lancet. 1983;1:1151–1155. - PubMed
-
- Depienne C., Stevanin G., Brice A., Durr A. Hereditary spastic paraplegias: An update. Curr. Opin. Neurol. 2007;20:674–680. - PubMed
-
- Fink J.K. Hereditary spastic paraplegia. Curr. Neurol. Neurosci. Rep. 2006;6:65–76. - PubMed
-
- Fink J.K. Advances in the hereditary spastic paraplegias. Exp. Neurol. 2003;184(Suppl 1):S106–S110. - PubMed
-
- Hazan J., Fonknechten N., Mavel D., Paternotte C., Samson D., Artiguenave F., Davoine C.S., Cruaud C., Dürr A., Wincker P. Spastin, a new AAA protein, is altered in the most frequent form of autosomal dominant spastic paraplegia. Nat. Genet. 1999;23:296–303. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
