

The genome-wide patterns of variation expose significant substructure in a founder population
- PMID: 19061986
- PMCID: PMC2668058
- DOI: 10.1016/j.ajhg.2008.11.005
The genome-wide patterns of variation expose significant substructure in a founder population
Abstract
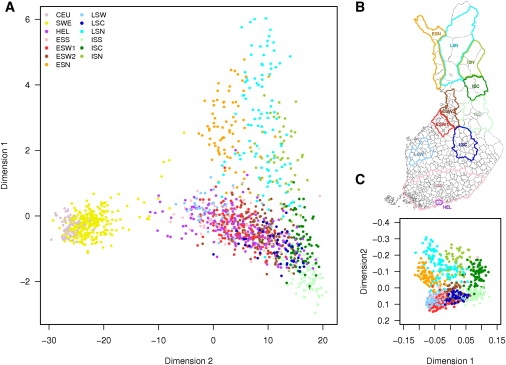
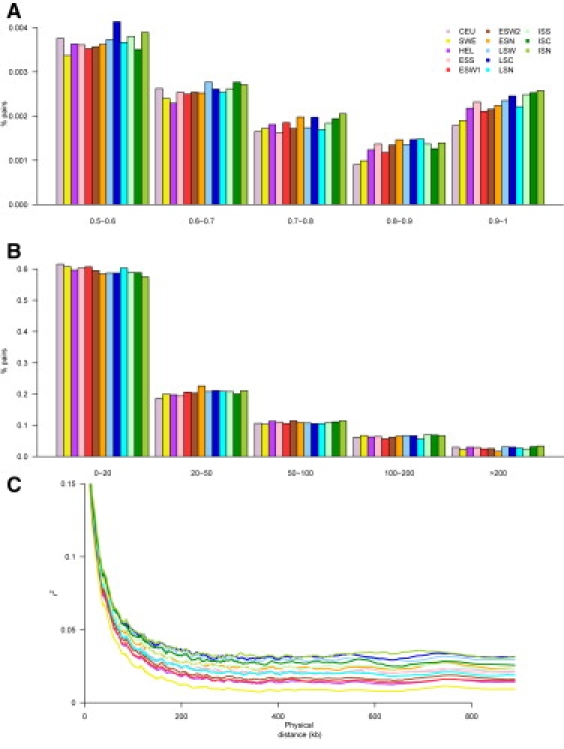
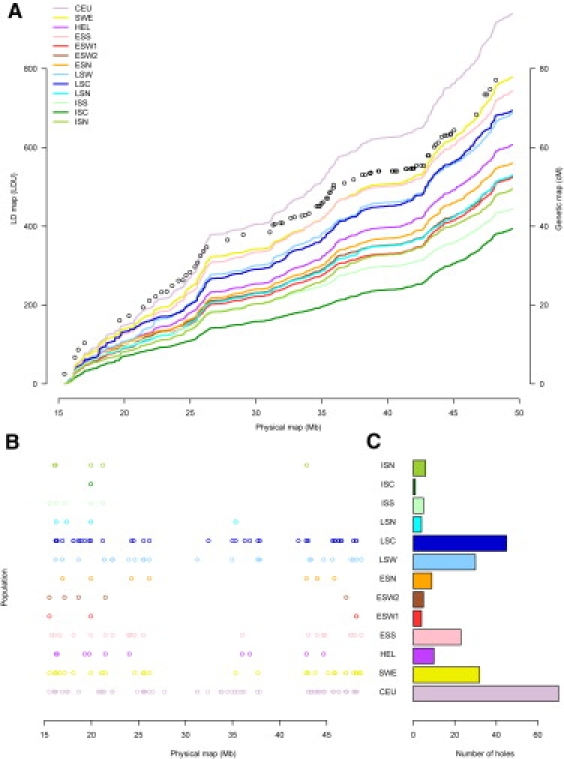
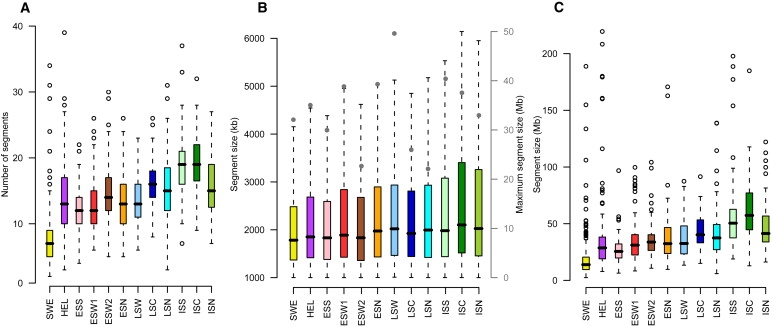
Although high-density SNP genotyping platforms generate a momentum for detailed genome-wide association (GWA) studies, an offshoot is a new insight into population genetics. Here, we present an example in one of the best-known founder populations by scrutinizing ten distinct Finnish early- and late-settlement subpopulations. By determining genetic distances, homozygosity, and patterns of linkage disequilibrium, we demonstrate that population substructure, and even individual ancestry, is detectable at a very high resolution and supports the concept of multiple historical bottlenecks resulting from consecutive founder effects. Given that genetic studies are currently aiming at identifying smaller and smaller genetic effects, recognizing and controlling for population substructure even at this fine level becomes imperative to avoid confounding and spurious associations. This study provides an example of the power of GWA data sets to demonstrate stratification caused by population history even within a seemingly homogeneous population, like the Finns. Further, the results provide interesting lessons concerning the impact of population history on the genome landscape of humans, as well as approaches to identify rare variants enriched in these subpopulations.
Figures
References
-
- Jakobsson M., Scholz S.W., Scheet P., Gibbs J.R., VanLiere J.M., Fung H.C., Szpiech Z.A., Degnan J.H., Wang K., Guerreiro R. Genotype, haplotype and copy-number variation in worldwide human populations. Nature. 2008;451:998–1003. - PubMed
-
- Li J.Z., Absher D.M., Tang H., Southwick A.M., Casto A.M., Ramachandran S., Cann H.M., Barsh G.S., Feldman M., Cavalli-Sforza L.L. Worldwide human relationships inferred from genome-wide patterns of variation. Science. 2008;319:1100–1104. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
