

Cis-regulatory variation is typically polyallelic in Drosophila
- PMID: 19064705
- PMCID: PMC2644954
- DOI: 10.1534/genetics.108.098459
Cis-regulatory variation is typically polyallelic in Drosophila
Abstract
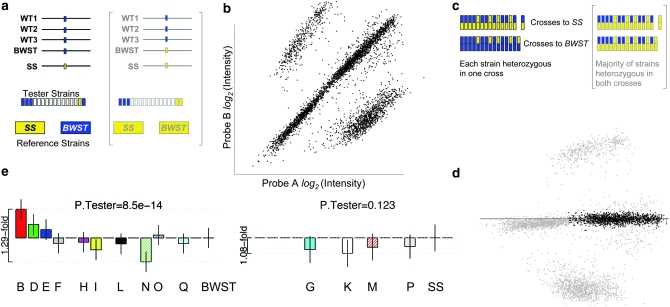
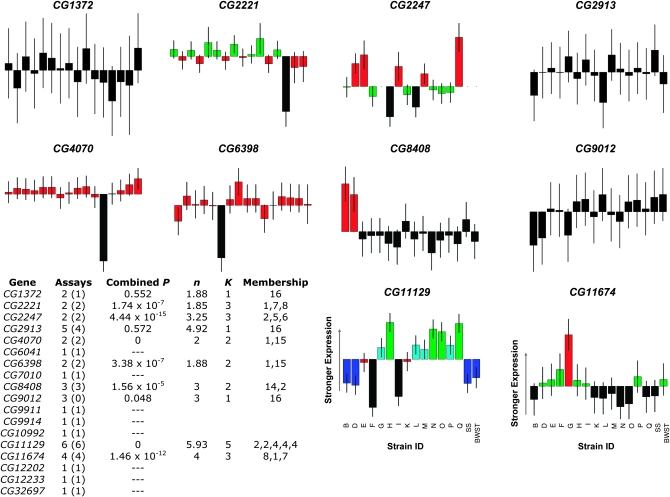
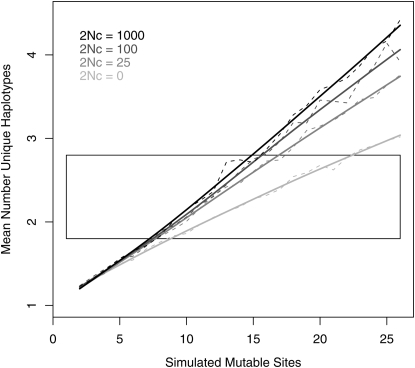
Gene expression levels vary heritably, with approximately 25-35% of the loci affecting expression acting in cis. We characterized standing cis-regulatory variation among 16 wild-derived strains of Drosophila melanogaster. Our experiment's robust biological and technical replication enabled precise estimates of variation in allelic expression on a high-throughput SNP genotyping platform. We observed concordant, significant differential allelic expression (DAE) in 7/10 genes queried with multiple SNPs, and every member of a set of eight additional, one-assay genes suggest significant DAE. Four of the high-confidence, multiple-assay genes harbor three or more statistically distinguishable allelic classes, often at intermediate frequency. Numerous intermediate-frequency, detectable regulatory polymorphisms cast doubt on a model in which cis-acting variation is a product of deleterious mutations of large effect. Comparing our data to predictions of population genetics theory using coalescent simulations, we estimate that a typical gene harbors 7-15 cis-regulatory sites (nucleotides) at which a selectively neutral mutation would elicit an observable expression phenotype. If standing cis-regulatory variation is actually slightly deleterious, the true mutational target size is larger.
Figures
References
-
- Andolfatto, P., 2005. Adaptive evolution of non-coding DNA in Drosophila. Nature 437 1149–1152. - PubMed
-
- Bray, N. J., and M. C. O'Donovan, 2006. Investigating cis-acting regulatory variation using assays of relative allelic expression. Psychiatr. Genet. 16 173–177. - PubMed
-
- Bray, N., P. Buckland, M. Owen and M. O'Donovan, 2003. Cis-acting variation in the expression of a high proportion of genes in human brain. Hum. Genet. 113 149–153. - PubMed
-
- Brem, R., G. Yvert, R. Clinton and L. Kruglyak, 2002. Genetic dissection of transcriptional regulation in budding yeast. Science 296 752–755. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
