

Expression of an activating mutation in the gene encoding the KATP channel subunit Kir6.2 in mouse pancreatic beta cells recapitulates neonatal diabetes
- PMID: 19065048
- PMCID: PMC2613450
- DOI: 10.1172/JCI35772
Expression of an activating mutation in the gene encoding the KATP channel subunit Kir6.2 in mouse pancreatic beta cells recapitulates neonatal diabetes
Abstract
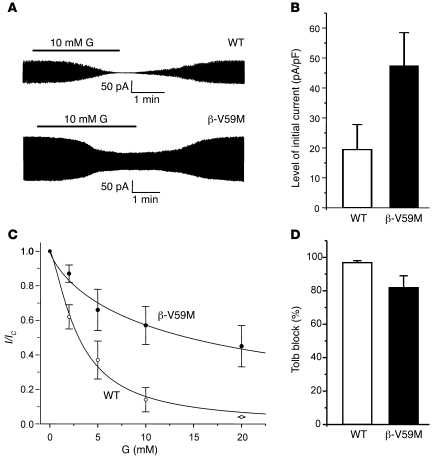
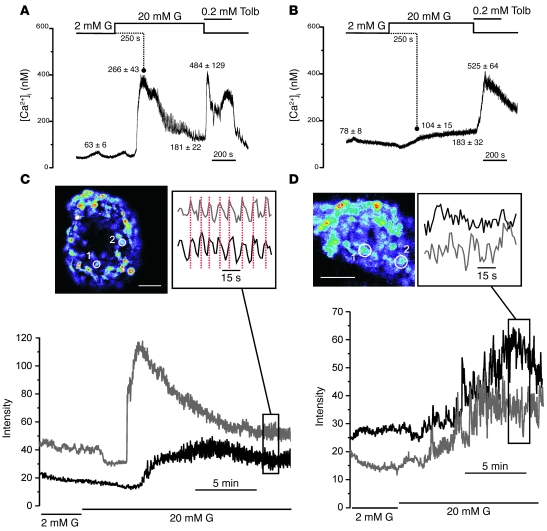
Neonatal diabetes is a rare monogenic form of diabetes that usually presents within the first six months of life. It is commonly caused by gain-of-function mutations in the genes encoding the Kir6.2 and SUR1 subunits of the plasmalemmal ATP-sensitive K+ (KATP) channel. To better understand this disease, we generated a mouse expressing a Kir6.2 mutation (V59M) that causes neonatal diabetes in humans and we used Cre-lox technology to express the mutation specifically in pancreatic beta cells. These beta-V59M mice developed severe diabetes soon after birth, and by 5 weeks of age, blood glucose levels were markedly increased and insulin was undetectable. Islets isolated from beta-V59M mice secreted substantially less insulin and showed a smaller increase in intracellular calcium in response to glucose. This was due to a reduced sensitivity of KATP channels in pancreatic beta cells to inhibition by ATP or glucose. In contrast, the sulfonylurea tolbutamide, a specific blocker of KATP channels, closed KATP channels, elevated intracellular calcium levels, and stimulated insulin release in beta-V59M beta cells, indicating that events downstream of KATP channel closure remained intact. Expression of the V59M Kir6.2 mutation in pancreatic beta cells alone is thus sufficient to recapitulate the neonatal diabetes observed in humans. beta-V59M islets also displayed a reduced percentage of beta cells, abnormal morphology, lower insulin content, and decreased expression of Kir6.2, SUR1, and insulin mRNA. All these changes are expected to contribute to the diabetes of beta-V59M mice. Their cause requires further investigation.
Figures






References
-
- Flanagan S.E., Edghill E.L., Gloyn A.L., Ellard S., Hattersley A.T. Mutations in KCNJ11, which encodes Kir6.2, are a common cause of diabetes diagnosed in the first 6 months of life, with the phenotype determined by genotype. Diabetologia. 2006;49:1190–1197. doi: 10.1007/s00125-006-0246-z. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
