

Genetic considerations relevant to intracranial hemorrhage and brain arteriovenous malformations
- PMID: 19066109
- PMCID: PMC2640934
- DOI: 10.1007/978-3-211-09469-3_38
Genetic considerations relevant to intracranial hemorrhage and brain arteriovenous malformations
Abstract
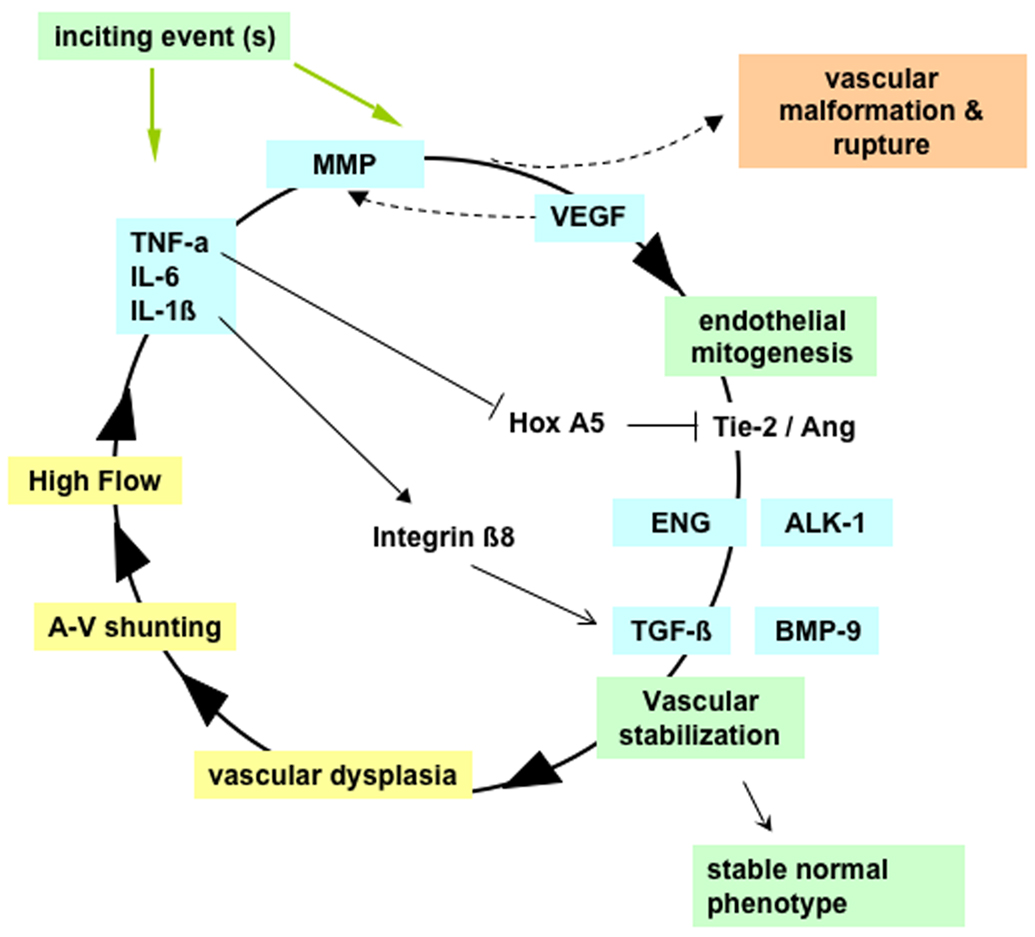
Brain arteriovenous malformations (AVMs) cause intracranial hemorrhage (ICH), especially in young adults. Molecular characterization of lesional tissue provides evidence for involvement of both angiogenic and inflammatory pathways, but the pathogenesis remains obscure and medical therapy is lacking. Abnormal expression patterns have been observed for proteins related to angiogenesis (e.g., vascular endothelial growth factor, angiopoietin-2, matrix metalloproteinase-9), and inflammation (e.g., interleukin-6 [IL-6] and myeloperoxidase). Macrophage and neutrophil invasion have also been observed in the absence of prior ICH. Candidate gene association studies have identified a number of germline variants associated with clinical ICH course and AVM susceptibility. A single nucleotide polymorphism (SNP) in activin receptor-like kinase-1 (ALK-1) is associated with AVM susceptibility, and SNPs in IL-6, tumor necrosis factor-alpha (TNF-alpha), and apolipoprotein-E (APOE) are associated with AVM rupture. These observations suggest that even without a complete understanding of the determinants of AVM development, the recent discoveries of downstream derangements in vascular function and integrity may offer potential targets for therapy development. Further, biomarkers can now be established for assessing ICH risk. These data will generate hypotheses that can be tested mechanistically in model systems, including surrogate phenotypes, such as vascular dysplasia and/or models recapitulating the clinical syndrome of recurrent spontaneous ICH.
Figures

References
-
- Achrol AS, Kim H, Pawlikowska L, Poon KY, Ko NU, McCulloch CE, Zaroff JG, Johnston SC, McDermott MW, Lawton MT, Kwok PY, Young WL. Association of tumor necrosis factor-alpha-238G>A and Apolipoprotein E2 polymorphisms with intracranial hemorrhage after brain arteriovenous malformation treatment. Neurosurgery. 2007;61:731–739. discussion 740. - PMC - PubMed
-
- Achrol AS, Pawlikowska L, McCulloch CE, Poon KY, Ha C, Zaroff JG, Johnston SC, Lee C, Lawton MT, Sidney S, Marchuk D, Kwok PY, Young WL. Tumor necrosis factor-alpha-238G>A promoter polymorphism is associated with increased risk of new hemorrhage in the natural course of patients with brain arteriovenous malformations. Stroke. 2006;37:231–234. - PubMed
-
- Arteriovenous Malformation Study Group. Arteriovenous malformations of the brain in adults. N Engl J Med. 1999;340:1812–1818. - PubMed
-
- Bayrak-Toydemir P, McDonald J, Akarsu N, Toydemir RM, Calderon F, Tuncali T, Tang W, Miller F, Mao R. A fourth locus for hereditary hemorrhagic telangiectasia maps to chromosome 7. Am J Med Genet A. 2006;140:2155–2162. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous