

Nephronophthisis
- PMID: 19066617
- PMCID: PMC2986221
- DOI: 10.1038/ejhg.2008.238
Nephronophthisis
Abstract
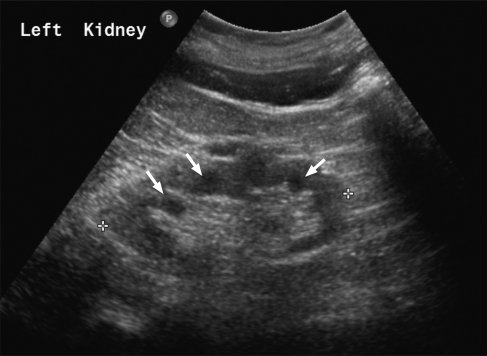
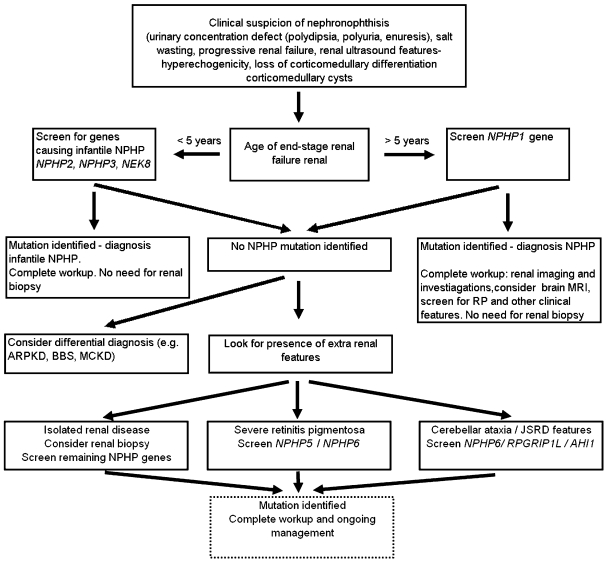
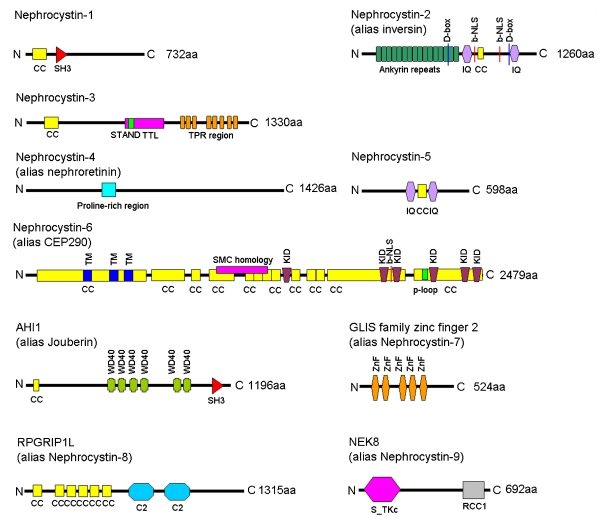
Nephronophthisis (NPHP) is an autosomal recessive kidney disorder characterized by chronic tubulointerstitial nephritis and leading to end-stage renal failure. NPHP as a renal entity is often part of a multisystem disorder and has been associated with many syndromes including Joubert syndrome (and related disorders) and Senior-Loken syndrome. Recent molecular genetic advances have allowed identification of several genes underlying NPHP. Most of these genes express their protein products, named nephrocystins, in primary cilial/basal body structures. Some nephrocystins are part of adherens junction and focal adhesion kinase protein complexes. This shared localization suggests that common pathogenic mechanisms within the kidney underlie this disease. Functional studies implicate nephrocystins in planar cell polarity pathways, which may be crucial for renal development and maintenance of tubular architecture.
Figures
References
-
- Hildebrandt F, Zhou W. Nephronophthisis-associated ciliopathies. J Am Soc Nephrol. 2007;18:1855–1871. - PubMed
-
- Zollinger HU, Mihatsch MJ, Edefonti A, Gaboardi F, Imbasciati E, Lennert T. Nephronophthisis (medullary cystic disease of the kidney). A study using electron microscopy, immunofluorescence, and a review of the morphological findings. Helv Paediatr Acta. 1980;35:509–530. - PubMed
-
- Krishnan R, Eley L, Sayer JA. Urinary concentration defects and mechanisms underlying nephronophthisis. Kidney Blood Press Res. 2008;31:152–162. - PubMed
-
- Gagnadoux MF, Bacri JL, Broyer M, Habib R. Infantile chronic tubulo-interstitial nephritis with cortical microcysts: variant of nephronophthisis or new disease entity. Pediatr Nephrol. 1989;3:50–55. - PubMed
-
- Parisi MA, Doherty D, Chance PF, Glass IA. Joubert syndrome (and related disorders) (OMIM 213300) Eur J Hum Genet. 2007;15:511–521. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
