

MEN1 gene and its mutations: basic and clinical implications
- PMID: 19068082
- PMCID: PMC11159991
- DOI: 10.1111/j.1349-7006.2008.01034.x
MEN1 gene and its mutations: basic and clinical implications
Abstract
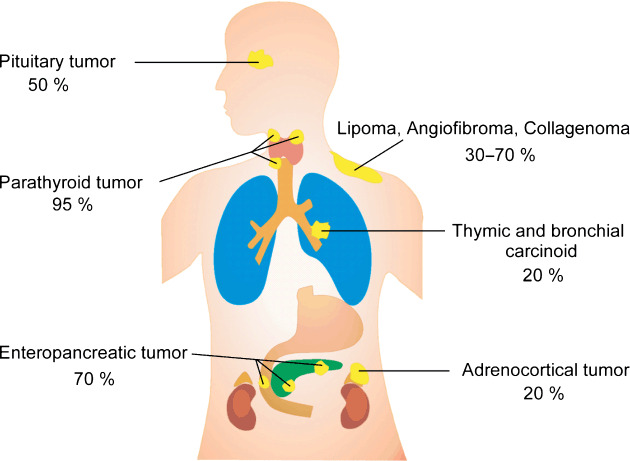
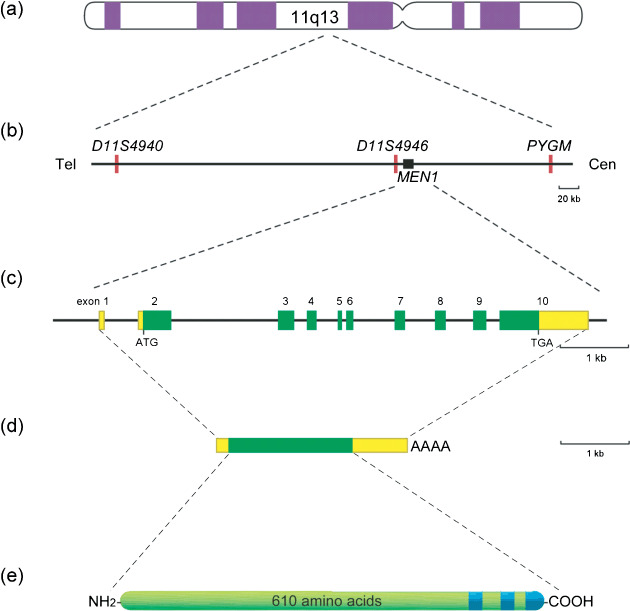
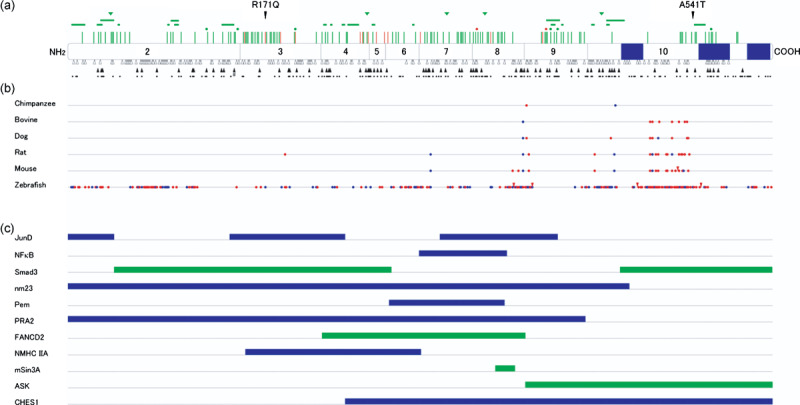
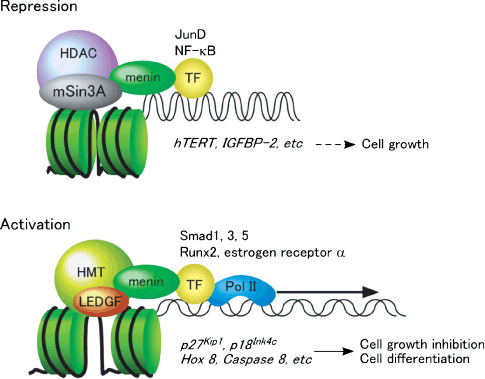
Heterozygous germline mutations of the tumor-suppressor gene MEN1 are responsible for multiple endocrine neoplasia type 1 (MEN1), a dominantly inherited familial cancer syndrome characterized by pituitary, parathyroid, and enteropancreatic tumors. Various mutations have been identified throughout the entire gene region in patients with MEN1 and related disorders. Neither mutation hot spot nor phenotype–genotype correlation has been established in MEN1 although some missense mutations may be specifically associated with a phenotype of familial isolated hyperparathyroidism. The gene product menin has been implicated in multiple roles, including gene transcription, maintenance of genomic integrity, and control of cell division and differentiation. These multiple functions are likely to be conferred by association with multiple protein factors. Occurrence of MEN1-causing missense mutations throughout menin also suggests the requirement of multiple binding factors for its full tumor-suppressive activity. The effect of menin depletion is highly tissue specific, but its underlying mechanism remains to be elucidated. A DNA test for MEN1 germline mutations is a useful tool for diagnosis of MEN1 although it needs further improvements
Figures
References
-
- Chandrasekharappa SC, Guru SC, Manickam P et al . Positional cloning of the gene for multiple endocrine neoplasia‐type 1. Science 1997; 276: 404–7. - PubMed
-
- Gardner DG. Recent advances in multiple endocrine neoplasia syndromes. Adv Intern Med 1997; 42: 597–627. - PubMed
-
- Brandi ML, Gagel RF, Angeli A et al . Guidelines for diagnosis and therapy of MEN type 1 and type 2. J Clin Endocrinol Metab 2001; 86: 5658–71. - PubMed
-
- Lemos MC, Thakker RV. Multiple endocrine neoplasia type 1 (MEN1): analysis of 1336 mutations reported in the first decade following identification of the gene. Hum Mutat 2008; 29: 22–32. - PubMed
-
- Balogh K, Hunyady L, Patocs A et al . MEN1 gene mutations in Hungarian patients with multiple endocrine neoplasia type 1. Clin Endocrinol 2007; 67: 727–34. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
