

Structural and dynamic study of the tetramerization region of non-erythroid alpha-spectrin: a frayed helix revealed by site-directed spin labeling electron paramagnetic resonance
- PMID: 19072330
- PMCID: PMC2649756
- DOI: 10.1021/bi8013032
Structural and dynamic study of the tetramerization region of non-erythroid alpha-spectrin: a frayed helix revealed by site-directed spin labeling electron paramagnetic resonance
Abstract
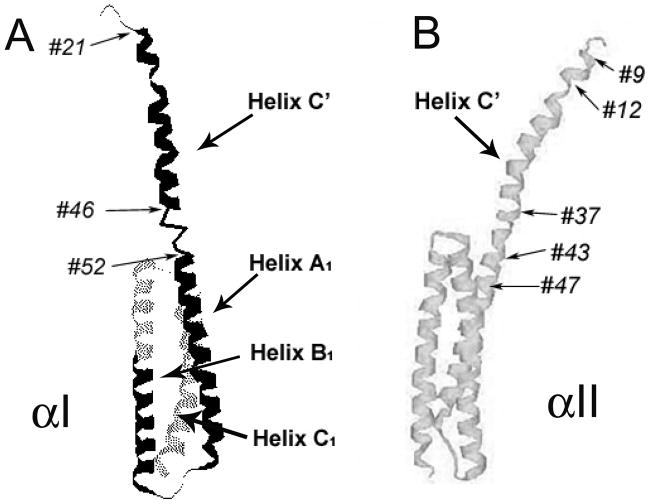
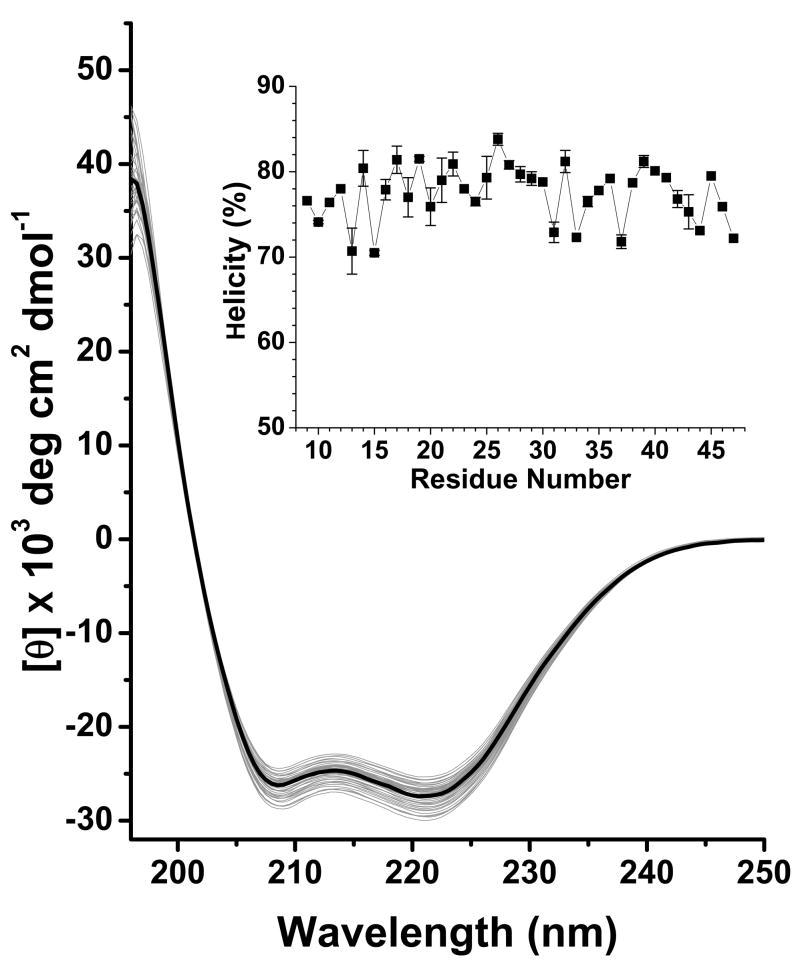
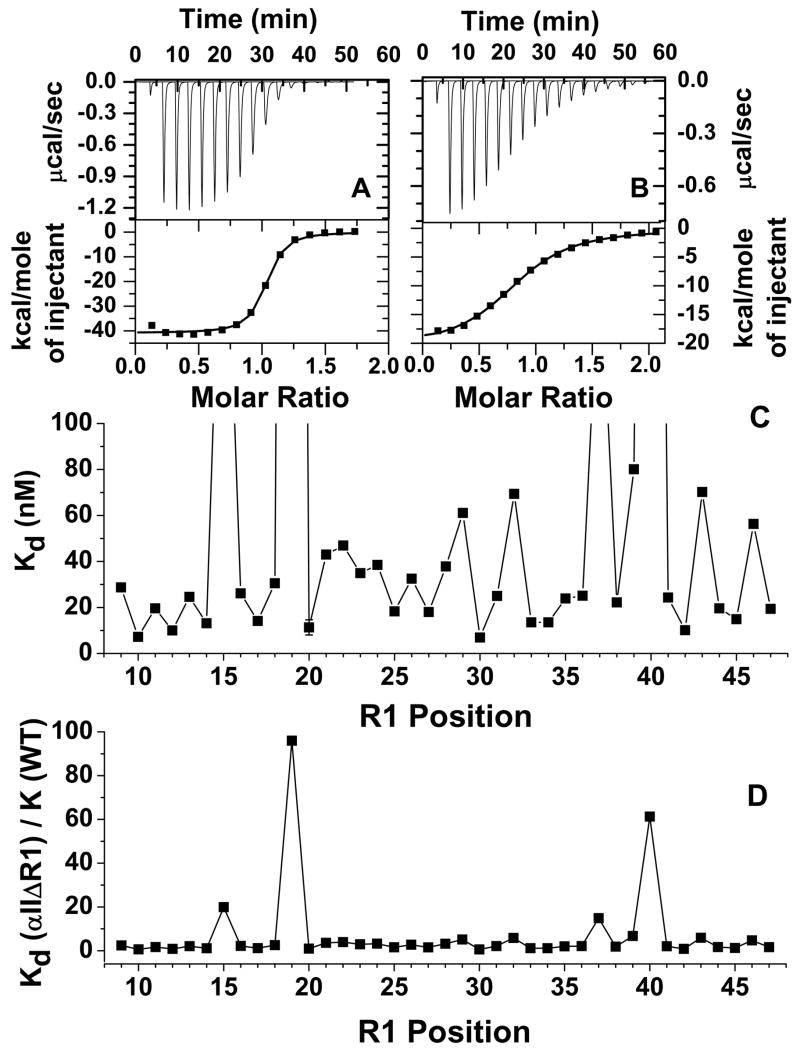
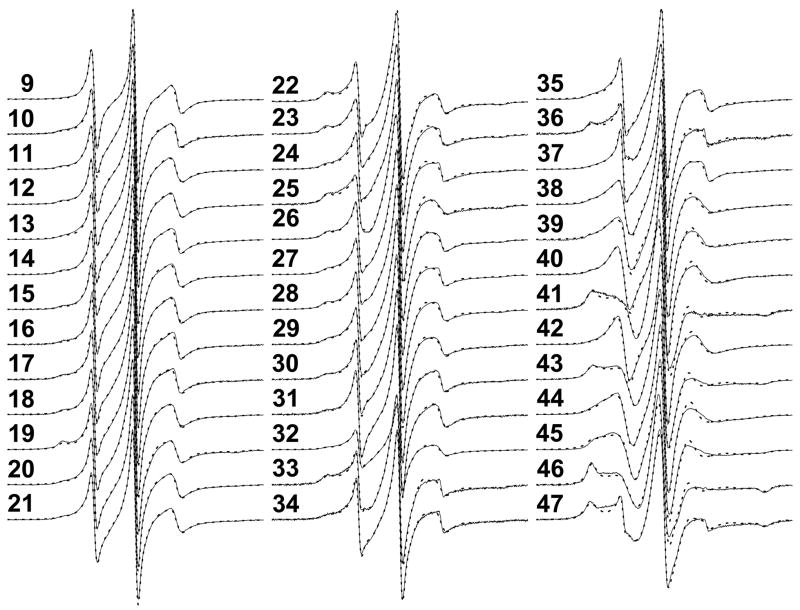
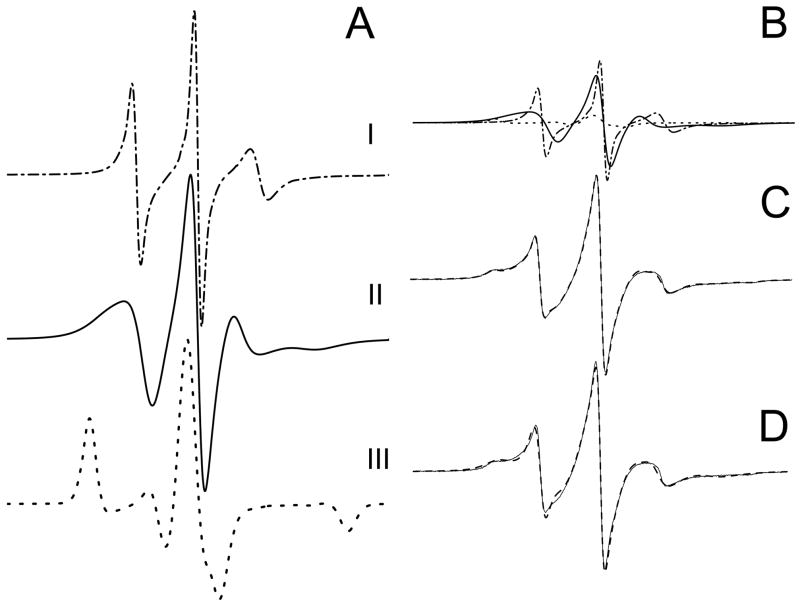
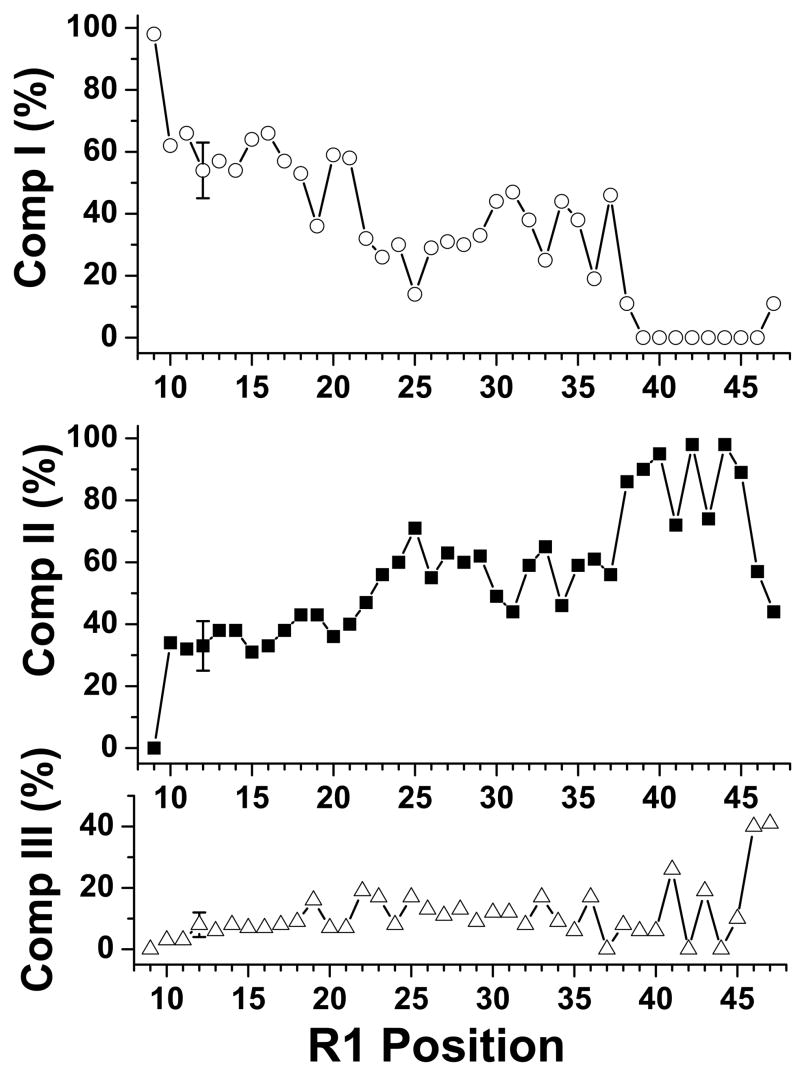
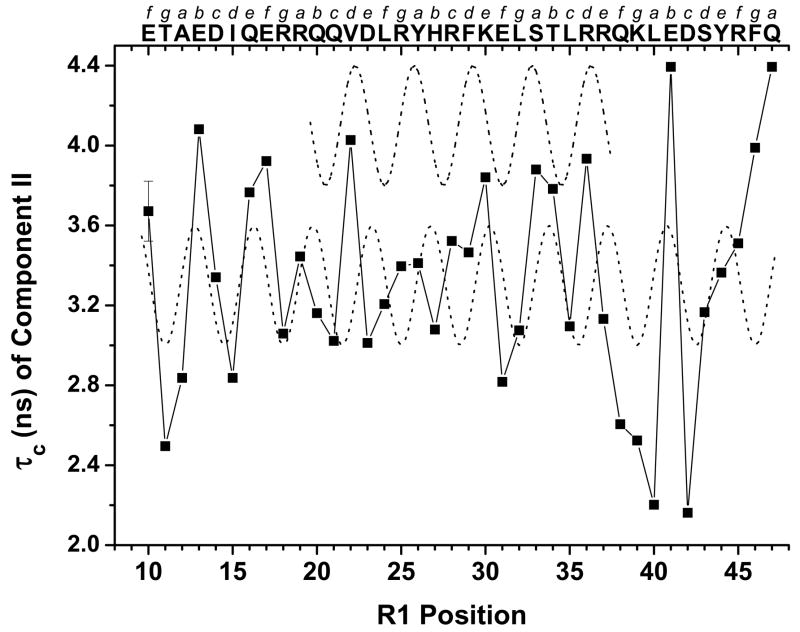
The N-terminal region of alpha-spectrin is responsible for its association with beta-spectrin in a heterodimer, forming functional tetramers. Non-erythroid alpha-spectrin (alphaII-spectrin) has a significantly higher association affinity for beta-spectrin than the homologous erythroid alpha-spectrin (alphaI-spectrin). We have previously determined the solution structure of the N-terminal region of alphaI-spectrin by NMR methods, but currently no structural information is available for alphaII-spectrin. We have used cysteine scanning, spin labeling electron paramagnetic resonance (EPR), and isothermal titration calorimetry (ITC) methods to study the tetramerization region of alphaII-spectrin. EPR data clearly show that, in alphaII-spectrin, the first nine N-terminal residues were unstructured, followed by an irregular helix (helix C'), frayed at the N-terminal end, but rigid at the C-terminal end, which merges into the putative triple-helical structural domain. The region corresponding to the important unstructured junction region linking helix C' to the first structural domain in alphaI-spectrin was clearly structured. On the basis of the published model for aligning helices A', B', and C', important interactions among residues in helix C' of alphaI- and alphaII-spectrin and helices A' and B' of betaI- and betaII-spectrin are identified, suggesting similar coiled coil helical bundling for spectrin I and II in forming tetramers. The differences in affinity are likely due to the differences in the conformation of the junction regions. Equilibrium dissociation constants of spin-labeled alphaII and betaI complexes from ITC measurements indicate that residues 15, 19, 37, and 40 are functionally important residues in alphaII-spectrin. Interestingly, all four corresponding homologous residues in alphaI-spectrin (residues 24, 28, 46, and 49) have been reported to be clinically significant residues involved in hematological diseases.
Figures
Similar articles
-
Crystal structure of the nonerythroid alpha-spectrin tetramerization site reveals differences between erythroid and nonerythroid spectrin tetramer formation.J Biol Chem. 2010 May 7;285(19):14572-84. doi: 10.1074/jbc.M109.080028. Epub 2010 Mar 14. J Biol Chem. 2010. PMID: 20228407 Free PMC article.
-
Apparent structural differences at the tetramerization region of erythroid and nonerythroid beta spectrin as discriminated by phage displayed scFvs.Protein Sci. 2011 May;20(5):867-79. doi: 10.1002/pro.617. Epub 2011 Mar 30. Protein Sci. 2011. PMID: 21412925 Free PMC article.
-
Conformational studies of the tetramerization site of human erythroid spectrin by cysteine-scanning spin-labeling EPR methods.Biochemistry. 2005 Dec 6;44(48):15898-905. doi: 10.1021/bi051009m. Biochemistry. 2005. PMID: 16313192
-
Conformational changes at the tetramerization site of erythroid alpha-spectrin upon binding beta-spectrin: a spin label EPR study.Biochemistry. 2008 Oct 7;47(40):10765-72. doi: 10.1021/bi800840p. Epub 2008 Sep 11. Biochemistry. 2008. PMID: 18783249 Free PMC article.
-
Spectrin and its interacting partners in nuclear structure and function.Exp Biol Med (Maywood). 2018 Mar;243(6):507-524. doi: 10.1177/1535370218763563. Exp Biol Med (Maywood). 2018. PMID: 29557213 Free PMC article. Review.
Cited by
-
Organization and dynamics of tryptophan residues in brain spectrin: novel insight into conformational flexibility.J Fluoresc. 2015 May;25(3):707-17. doi: 10.1007/s10895-015-1556-7. Epub 2015 Apr 3. J Fluoresc. 2015. PMID: 25835748
-
Expression, purification, and reconstitution of the voltage-sensing domain from Ci-VSP.Biochemistry. 2012 Oct 16;51(41):8132-42. doi: 10.1021/bi300980q. Epub 2012 Oct 5. Biochemistry. 2012. PMID: 22989304 Free PMC article.
-
Crystal structure of the nonerythroid alpha-spectrin tetramerization site reveals differences between erythroid and nonerythroid spectrin tetramer formation.J Biol Chem. 2010 May 7;285(19):14572-84. doi: 10.1074/jbc.M109.080028. Epub 2010 Mar 14. J Biol Chem. 2010. PMID: 20228407 Free PMC article.
-
Probing conformational stability and dynamics of erythroid and nonerythroid spectrin: effects of urea and guanidine hydrochloride.PLoS One. 2015 Jan 24;10(1):e0116991. doi: 10.1371/journal.pone.0116991. eCollection 2015. PLoS One. 2015. PMID: 25617632 Free PMC article.
-
Association studies of erythroid alpha-spectrin at the tetramerization site.Br J Haematol. 2009 Nov;147(3):392-5. doi: 10.1111/j.1365-2141.2009.07876.x. Epub 2009 Aug 31. Br J Haematol. 2009. PMID: 19747366 Free PMC article.
References
-
- Bennett V, Baines AJ. Spectrin and ankyrin-based pathways: metazoan inventions for integrating cells into tissues. Physiol Rev. 2001;81:1353–1392. - PubMed
-
- Speicher DW, DeSilva TM, Speicher KD, Ursitt JA, Hembach P, Weglarz L. Location of the human red cell spectrin tetramer binding site & detection of a related “closed” hairpin loop dimer using proteolytic footprinting. J Biol Chem. 1993;268:4227–4235. - PubMed
-
- DeSilva TM, Peng KC, Speicher KD, Speicher DW. Analysis of human red cell spectrin tetramer (head-to-head) assembly using complementary univalent peptides. Biochemistry. 1992;31:10872–10878. - PubMed
-
- Agre P. Clinical relevance of basic research on red cell membranes. Clin Res. 1992;40:176–186. - PubMed
-
- Delauna J, Dhermy D. Mutations involving the spectrin heterodimer contact site: clinical expression and alterations in specific function. Semin Hematol. 1993;30:21–33. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
