

Where asthma and hypersensitivity pneumonitis meet and differ: noneosinophilic severe asthma
- PMID: 19074616
- PMCID: PMC2631313
- DOI: 10.2353/ajpath.2009.071151
Where asthma and hypersensitivity pneumonitis meet and differ: noneosinophilic severe asthma
Abstract
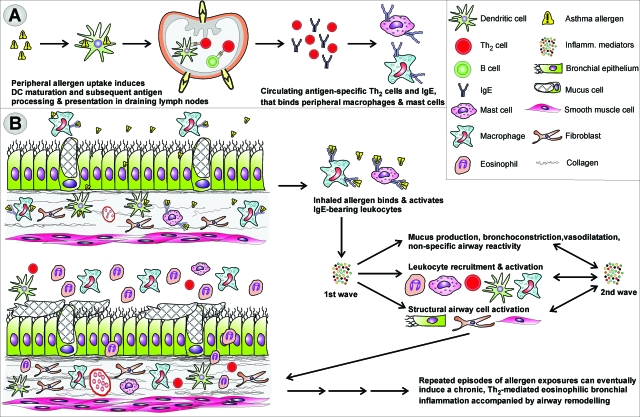
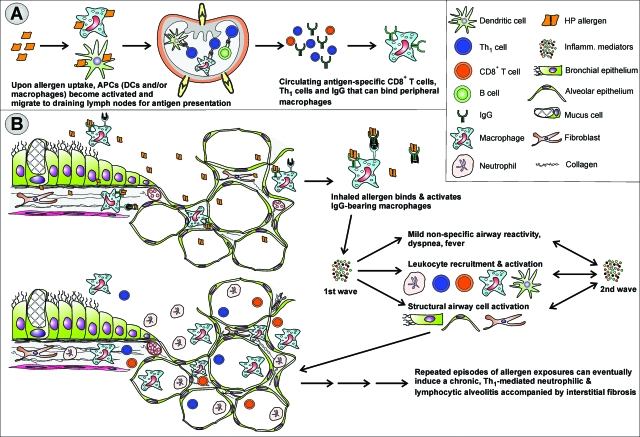
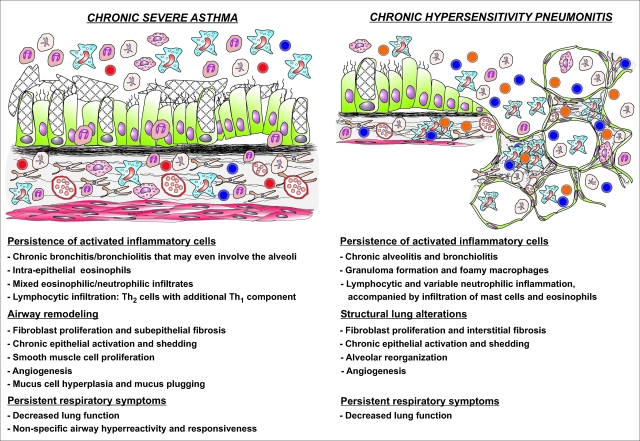
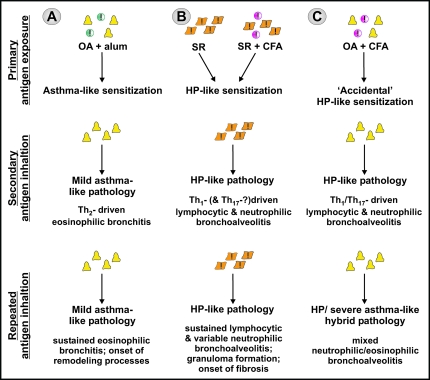
Asthma is a type-I allergic airway disease characterized by Th(2) cells and IgE. Episodes of bronchial inflammation, eosinophilic in nature and promoting bronchoconstriction, may become chronic and lead to persistent respiratory symptoms and irreversible structural airway changes. Representative mostly of mild to moderate asthma, this clinical definition fails to account for the atypical and often more severe phenotype found in a considerable proportion of asthmatics who have increased neutrophil cell counts in the airways as a distinguishing trait. Neutrophilic inflammation is a hallmark of another type of allergic airway pathology, hypersensitivity pneumonitis. Considered as an immune counterpart of asthma, hypersensitivity pneumonitis is a prototypical type-III allergic inflammatory reaction involving the alveoli and lung interstitium, steered by Th(1) cells and IgG and, in its chronic form, accompanied by fibrosis. Although pathologically very different and commonly approached as separate disorders, as discussed in this review, clinical studies as well as data from animal models reveal undeniable parallels between both airway diseases. Danger signaling elicited by the allergenic agent or by accompanying microbial patterns emerges as critical in enabling immune sensitization and in determining the type of sensitization and ensuing allergic disease. On this basis, we propose that asthma allergens cause severe noneosinophilic asthma because of sensitization in the presence of hypersensitivity pneumonitis-promoting danger signaling.
Figures
References
-
- Wills-Karp M, Santeliz J, Karp CL. The germless theory of allergic disease: revisiting the hygiene hypothesis. Nat Rev Immunol. 2001;1:69–75. - PubMed
-
- Herrick CA, Bottomly K. To respond or not to respond: T cells in allergic asthma. Nat Rev Immunol. 2003;3:405–412. - PubMed
-
- Tattersfield AE, Knox AJ, Britton JR, Hall IP. Asthma. Lancet. 2002;360:1313–1322. - PubMed
-
- Kleeberger SR, Peden D. Gene-environment interactions in asthma and other respiratory diseases. Annu Rev Med. 2005;56:383–400. - PubMed
-
- Bossé Y, Hudson TJ. Toward a comprehensive set of asthma susceptibility genes. Annu Rev Med. 2007;58:171–184. - PubMed
