

Protein kinase C-delta and phosphatidylinositol 3-kinase/Akt activate mammalian target of rapamycin to modulate NF-kappaB activation and intercellular adhesion molecule-1 (ICAM-1) expression in endothelial cells
- PMID: 19074768
- PMCID: PMC2640976
- DOI: 10.1074/jbc.M805032200
Protein kinase C-delta and phosphatidylinositol 3-kinase/Akt activate mammalian target of rapamycin to modulate NF-kappaB activation and intercellular adhesion molecule-1 (ICAM-1) expression in endothelial cells
Abstract
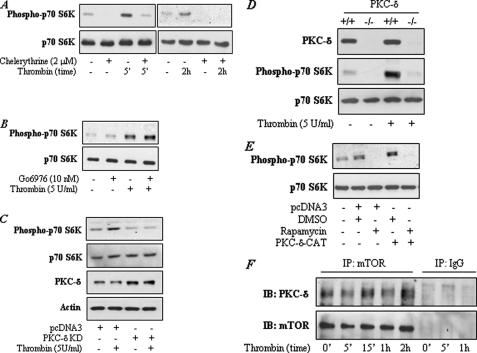
We have shown that the mammalian target of rapamycin (mTOR) down-regulates thrombin-induced ICAM-1 expression in endothelial cells by suppressing the activation of NF-kappaB. However, the mechanisms by which mTOR is activated to modulate these responses remain to be addressed. Here, we show that thrombin engages protein kinase C (PKC)-delta and phosphattidylinositol 3-kinase (PI3K)/Akt pathways to activate mTOR and thereby dampens NF-kappaB activation and intercellular adhesion molecule 1 (ICAM-1) expression. Stimulation of human vascular endothelial cells with thrombin induced the phosphorylation of mTOR and its downstream target p70 S6 kinase in a PKC-delta- and PI3K/Akt-dependent manner. Consistent with this, thrombin-induced phosphorylation of p70 S6 kinase was defective in embryonic fibroblasts from mice with targeted disruption of PKC-delta (Pkc-delta(-)(/)(-)), p85alpha and p85beta subunits of the PI3K (p85alpha(-)(/)(-)beta(-)(/)(-)), or Akt1 and Akt2 (Akt1(-)(/)(-)2(-)(/)(-)). Furthermore, we observed that expression of the constitutively active form of PKC-delta or Akt was sufficient to induce NF-kappaB activation and ICAM-1 expression, and that co-expression of mTOR suppressed these responses. In reciprocal experiments, inhibition/depletion of mTOR augmented NF-kappaB activation and ICAM-1 expression induced by PKC-delta or Akt. In control experiments, increasing or impairing mTOR signaling by the above approaches produced similar effects on NF-kappaB activation and ICAM-1 expression induced by thrombin. Thus, these data reveal an important role of PKC-delta and PI3K/Akt pathways in activating mTOR as an endogenous modulator to ensure a tight regulation of NF-kappaB signaling of ICAM-1 expression in endothelial cells.
Figures







References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
