

ESE-1/EGR-1 pathway plays a role in tolfenamic acid-induced apoptosis in colorectal cancer cells
- PMID: 19074849
- PMCID: PMC2643071
- DOI: 10.1158/1535-7163.MCT-08-0548
ESE-1/EGR-1 pathway plays a role in tolfenamic acid-induced apoptosis in colorectal cancer cells
Abstract
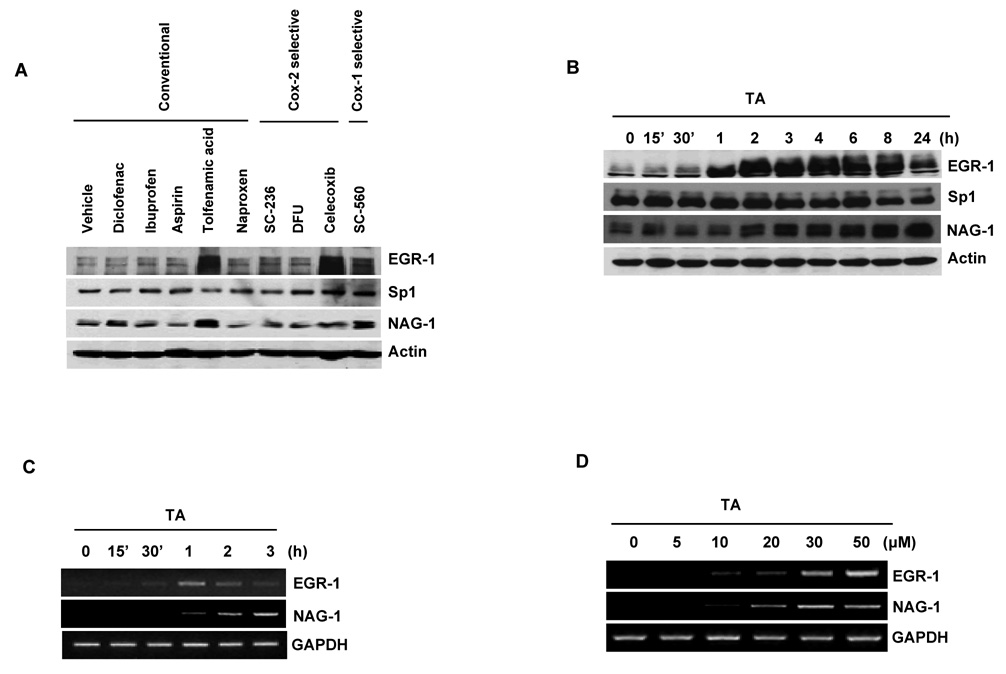
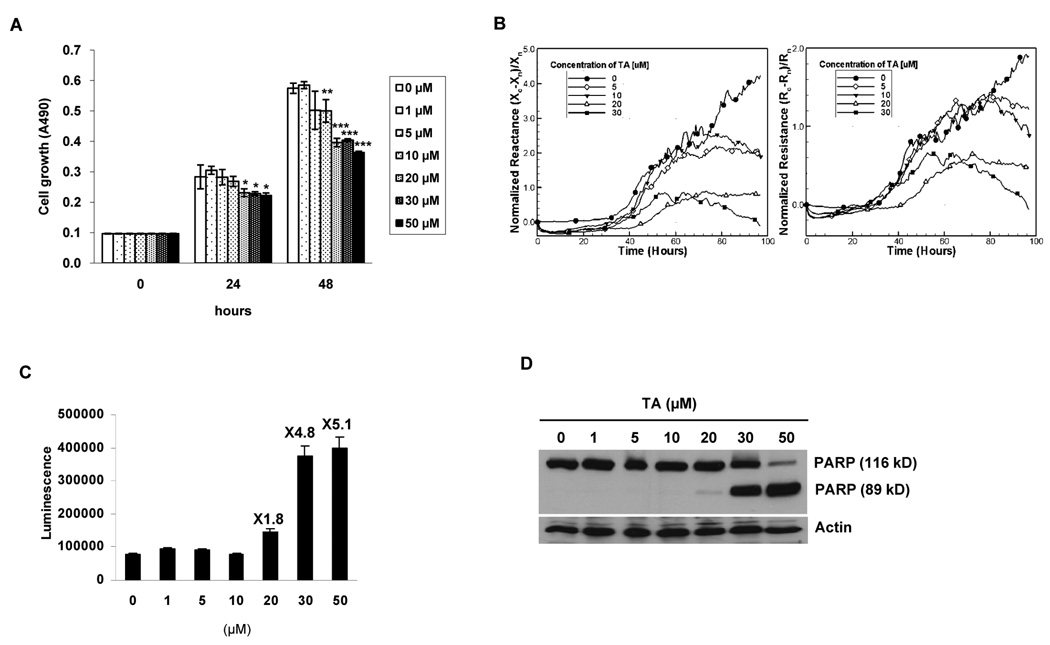
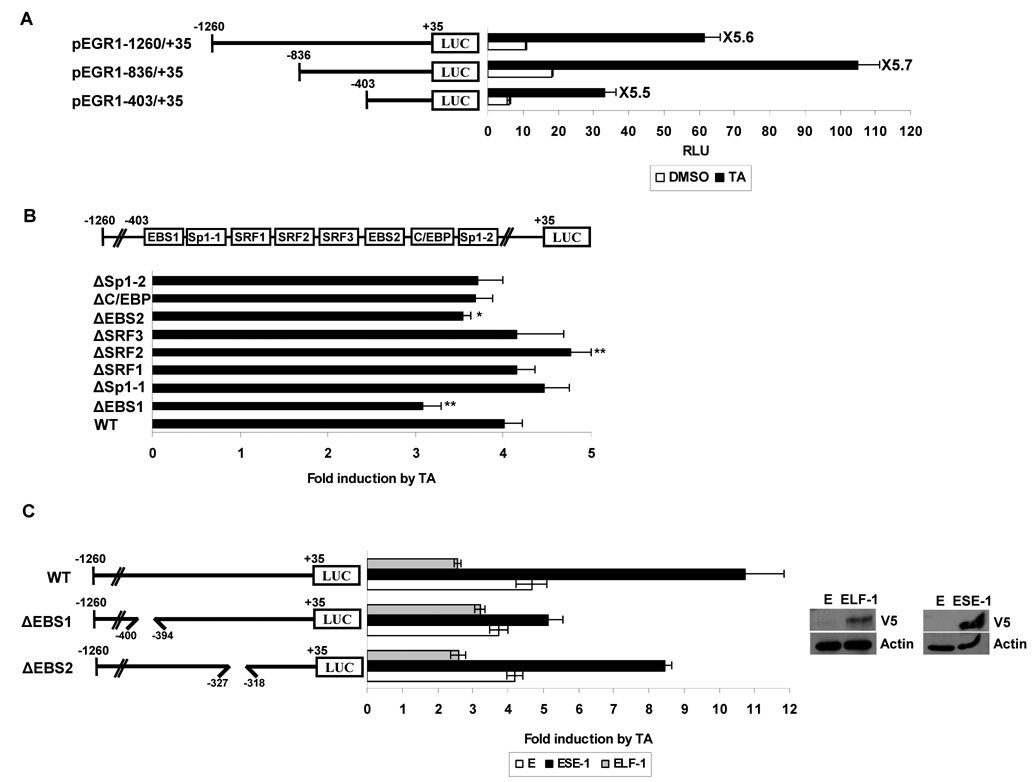
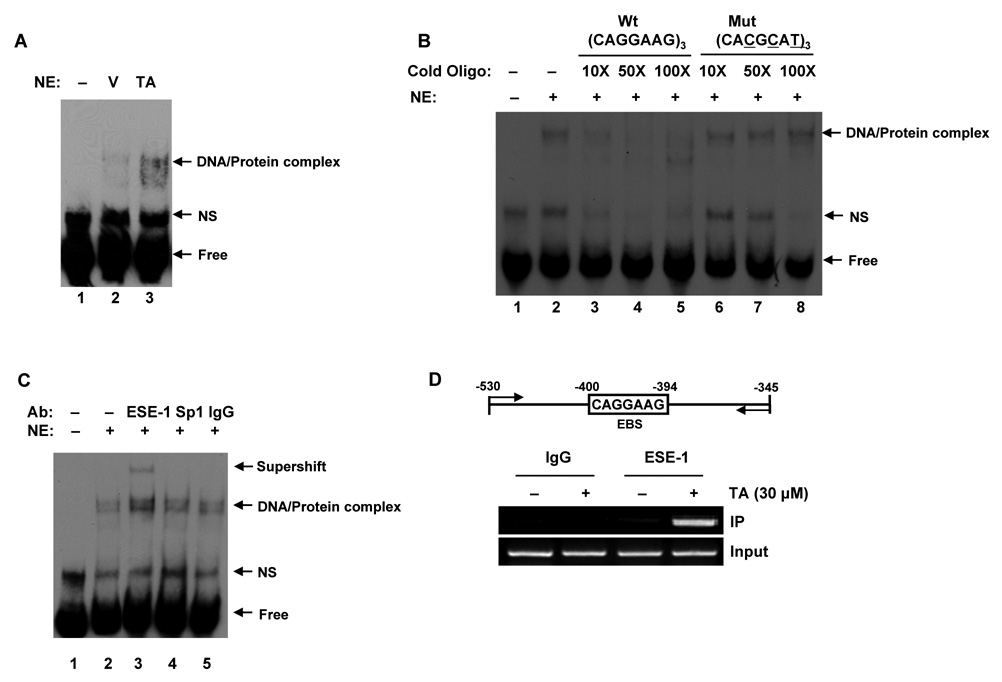
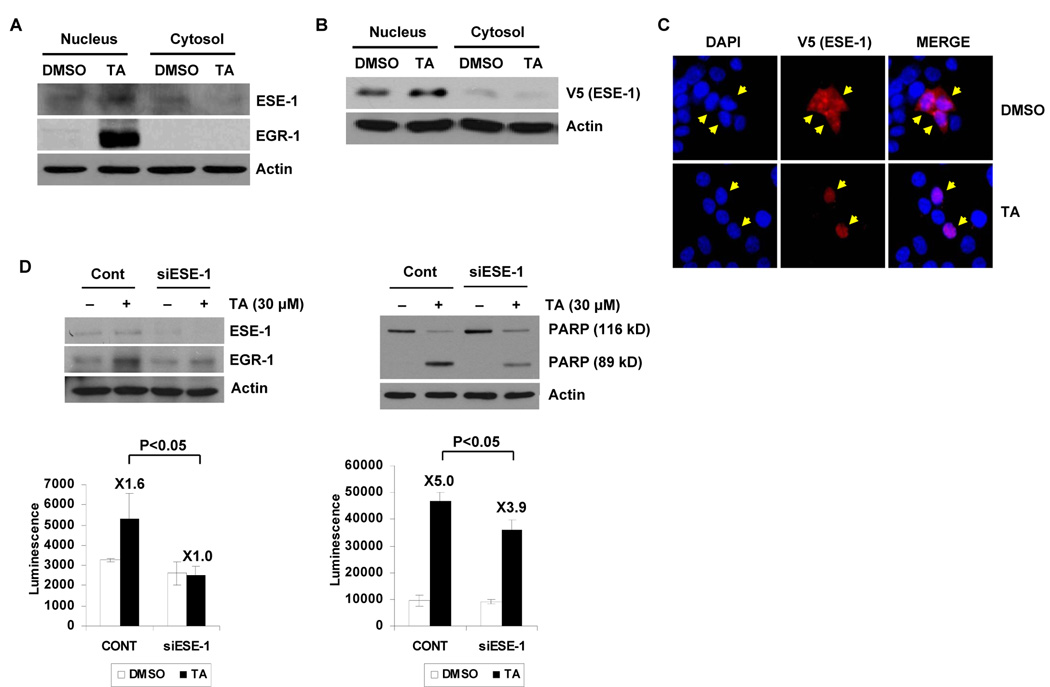
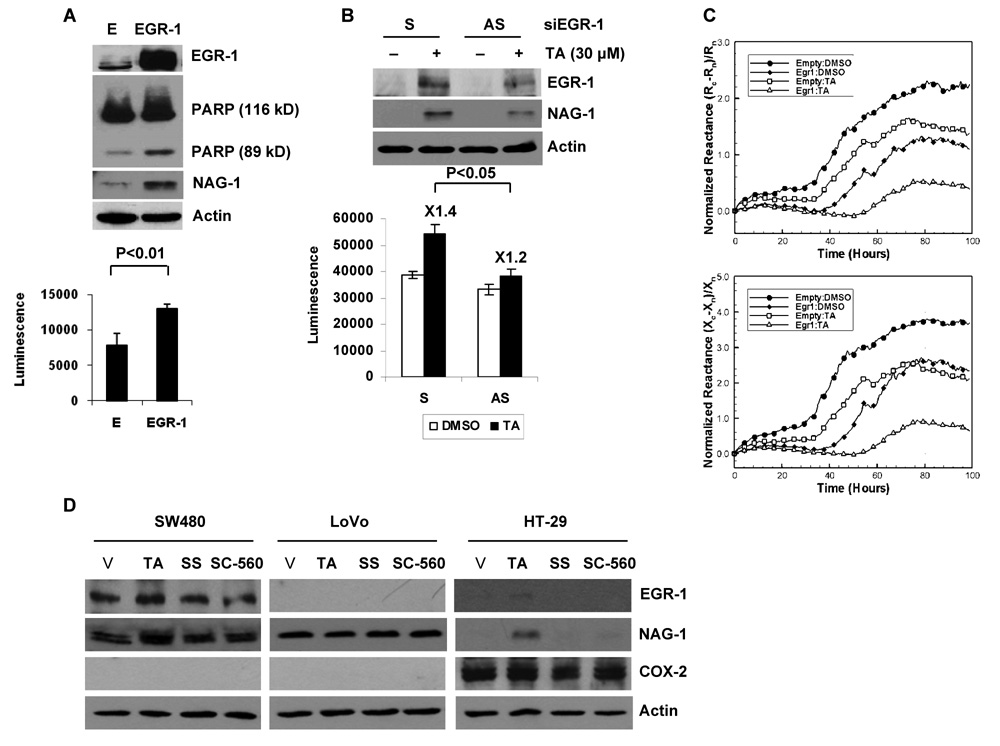
Nonsteroidal anti-inflammatory drugs (NSAIDs) are known to prevent colorectal tumorigenesis. Although antitumor effects of NSAIDs are mainly due to inhibition of cyclooxygenase activity, there is increasing evidence that cyclooxygenase-independent mechanisms may also play an important role. The early growth response-1 (EGR-1) gene is a member of the immediate-early gene family and has been identified as a tumor suppressor gene. Tolfenamic acid is a NSAID that exhibits anticancer activity in a pancreatic cancer model. In the present study, we investigated the anticancer activity of tolfenamic acid in human colorectal cancer cells. Tolfenamic acid treatment inhibited cell growth and induced apoptosis as measured by caspase activity and bioelectric impedance. Tolfenamic acid induced EGR-1 expression at the transcription level, and analysis of the EGR-1 promoter showed that a putative ETS-binding site, located at -400 and -394 bp, was required for activation by tolfenamic acid. The electrophoretic mobility shift assay and chromatin immunoprecipitation assay confirmed that this sequence specifically bound to the ETS family protein epithelial-specific ETS-1 (ESE-1) transcription factor. Tolfenamic acid also facilitated translocation of endogenous and exogenous ESE-1 to the nucleus in colorectal cancer cells, and gene silencing using ESE-1 small interfering RNA attenuated tolfenamic acid-induced EGR-1 expression and apoptosis. Overexpression of EGR-1 increased apoptosis and decreased bioelectrical impedance, and silencing of endogenous EGR-1 prevented tolfenamic acid-induced apoptosis. These results show that activation of ESE-1 via enhanced nuclear translocation mediates tolfenamic acid-induced EGR-1 expression, which plays a critical role in the activation of apoptosis.
Figures
References
-
- Huls G, Koornstra JJ, Kleibeuker JH. Non-steroidal anti-inflammatory drugs and molecular carcinogenesis of colorectal carcinomas. Lancet. 2003;362:230–232. - PubMed
-
- Gupta RA, Dubois RN. Colorectal cancer prevention and treatment by inhibition of cyclooxygenase-2. Nat Rev Cancer. 2001;1:11–21. - PubMed
-
- Grosch S, Tegeder I, Niederberger E, Brautigam L, Geisslinger G. COX-2 independent induction of cell cycle arrest and apoptosis in colon cancer cells by the selective COX-2 inhibitor celecoxib. Faseb J. 2001;15:2742–2744. - PubMed
-
- Leahy KM, Ornberg RL, Wang Y, Zweifel BS, Koki AT, Masferrer JL. Cyclooxygenase-2 inhibition by celecoxib reduces proliferation and induces apoptosis in angiogenic endothelial cells in vivo. Cancer Res. 2002;62:625–631. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
