

250K single nucleotide polymorphism array karyotyping identifies acquired uniparental disomy and homozygous mutations, including novel missense substitutions of c-Cbl, in myeloid malignancies
- PMID: 19074904
- PMCID: PMC2668538
- DOI: 10.1158/0008-5472.CAN-08-2754
250K single nucleotide polymorphism array karyotyping identifies acquired uniparental disomy and homozygous mutations, including novel missense substitutions of c-Cbl, in myeloid malignancies
Abstract
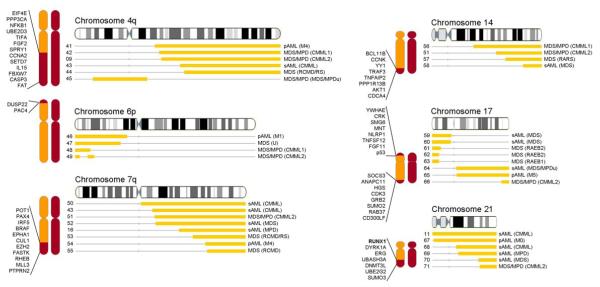
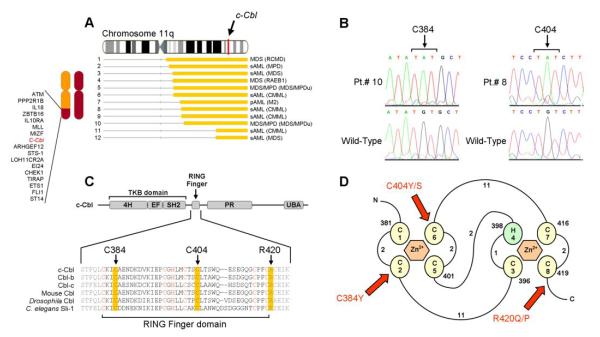
Two types of acquired loss of heterozygosity are possible in cancer: deletions and copy-neutral uniparental disomy (UPD). Conventionally, copy number losses are identified using metaphase cytogenetics, whereas detection of UPD is accomplished by microsatellite and copy number analysis and as such, is not often used clinically. Recently, introduction of single nucleotide polymorphism (SNP) microarrays has allowed for the systematic and sensitive detection of UPD in hematologic malignancies and other cancers. In this study, we have applied 250K SNP array technology to detect previously cryptic chromosomal changes, particularly UPD, in a cohort of 301 patients with myelodysplastic syndromes (MDS), overlap MDS/myeloproliferative disorders (MPD), MPD, and acute myeloid leukemia. We show that UPD is a common chromosomal defect in myeloid malignancies, particularly in chronic myelomonocytic leukemia (CMML; 48%) and MDS/MPD-unclassifiable (38%). Furthermore, we show that mapping minimally overlapping segmental UPD regions can help target the search for both known and unknown pathogenic mutations, including newly identified missense mutations in the proto-oncogene c-Cbl in 7 of 12 patients with UPD11q. Acquired mutations of c-Cbl E3 ubiquitin ligase may explain the pathogenesis of a clonal process in a subset of MDS/MPD, including CMML.
Figures


References
-
- Robinson WP. Mechanisms leading to uniparental disomy and their clinical consequences. Bioessays. 2000;22:452–9. - PubMed
-
- Jiang YH, Bressler J, Beaudet AL. Epigenetics and human disease. Annu.Rev.Genomics Hum.Genet. 2004;5:479–510. - PubMed
-
- Kralovics R, Guan Y, Prchal JT. Acquired uniparental disomy of chromosome 9p is a frequent stem cell defect in polycythemia vera. Exp.Hematol. 2002;30:229–36. - PubMed
-
- Baxter EJ, Scott LM, Campbell PJ, et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet. 2005;365:1054–61. - PubMed
-
- Kralovics R, Passamonti F, Buser AS, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med. 2005;352:1779–90. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
