

IL-17A and IL-17F do not contribute vitally to autoimmune neuro-inflammation in mice
- PMID: 19075395
- PMCID: PMC2613466
- DOI: 10.1172/JCI35997
IL-17A and IL-17F do not contribute vitally to autoimmune neuro-inflammation in mice
Abstract
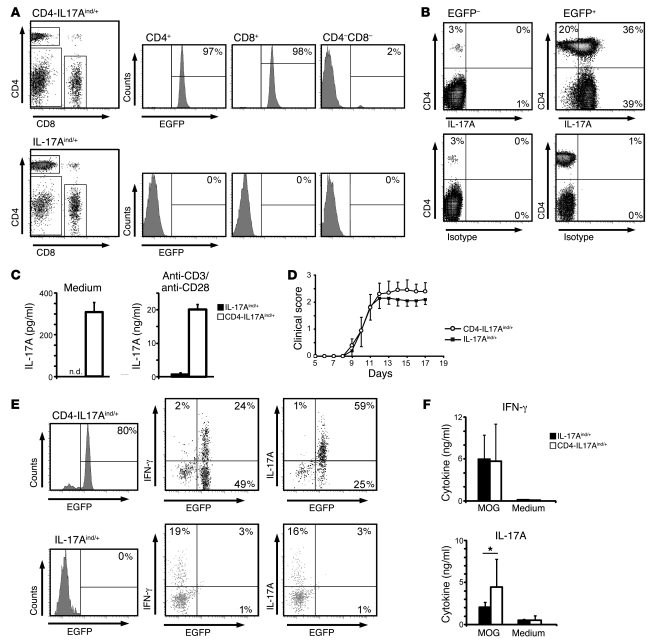
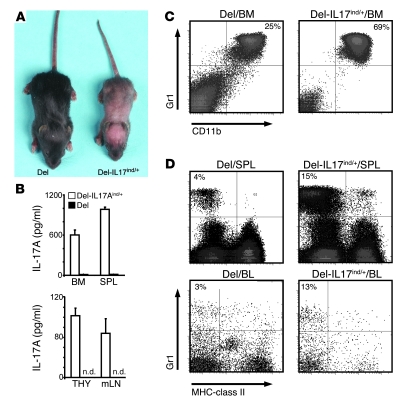
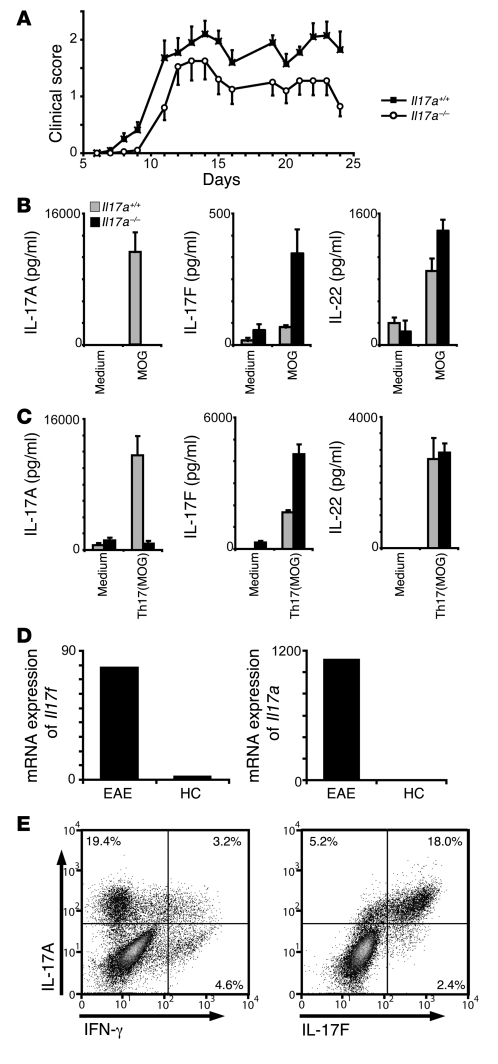
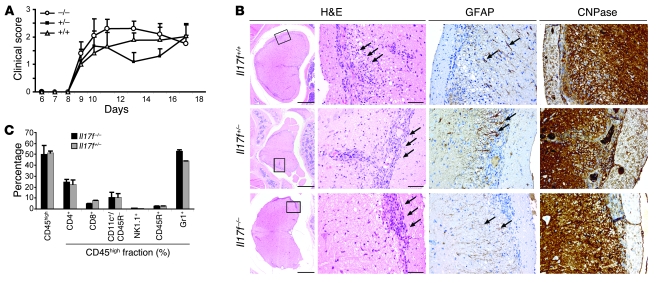
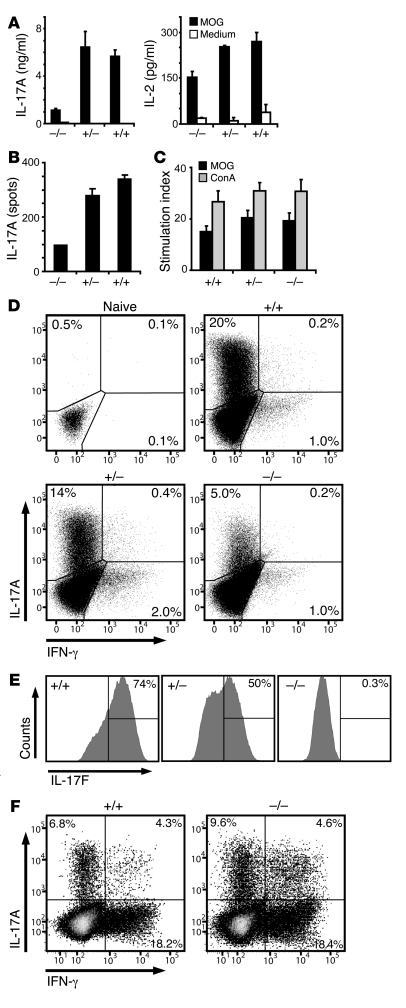
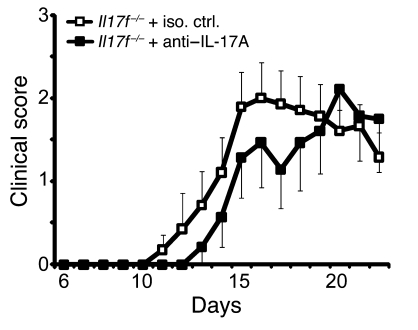
The clear association of Th17 cells with autoimmune pathogenicity implicates Th17 cytokines as critical mediators of chronic autoimmune diseases such as EAE. To study the impact of IL-17A on CNS inflammation, we generated transgenic mice in which high levels of expression of IL-17A could be initiated after Cre-mediated recombination. Although ubiquitous overexpression of IL-17A led to skin inflammation and granulocytosis, T cell-specific IL-17A overexpression did not have a perceptible impact on the development and health of the mice. In the context of EAE, neither the T cell-driven overexpression of IL-17A nor its complete loss had a major impact on the development of clinical disease. Since IL-17F may be able to compensate for the loss of IL-17A, we also generated IL-17F-deficient mice. This strain was fully susceptible to EAE and displayed unaltered emergence and expansion of autoreactive T cells during disease. To eliminate potential compensatory effects of either cytokine, we treated IL-17F-deficient mice with antagonistic monoclonal antibodies specific for IL-17A and found again only a minimal beneficial impact on disease development. We conclude therefore that both IL-17A and IL-17F, while prominently expressed by an encephalitogenic T cell population, may only marginally contribute to the development of autoimmune CNS disease.
Figures
References
-
- Willenborg D.O., Fordham S., Bernard C.C., Cowden W.B., Ramshaw I.A. IFN-gamma plays a critical down-regulatory role in the induction and effector phase of myelin oligodendrocyte glycoprotein-induced autoimmune encephalomyelitis. J. Immunol. 1996;157:3223–3227. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
