

Brain glucose hypometabolism and oxidative stress in preclinical Alzheimer's disease
- PMID: 19076441
- PMCID: PMC2661241
- DOI: 10.1196/annals.1427.007
Brain glucose hypometabolism and oxidative stress in preclinical Alzheimer's disease
Abstract
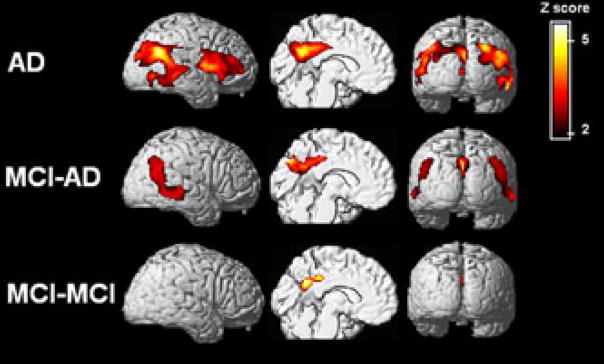
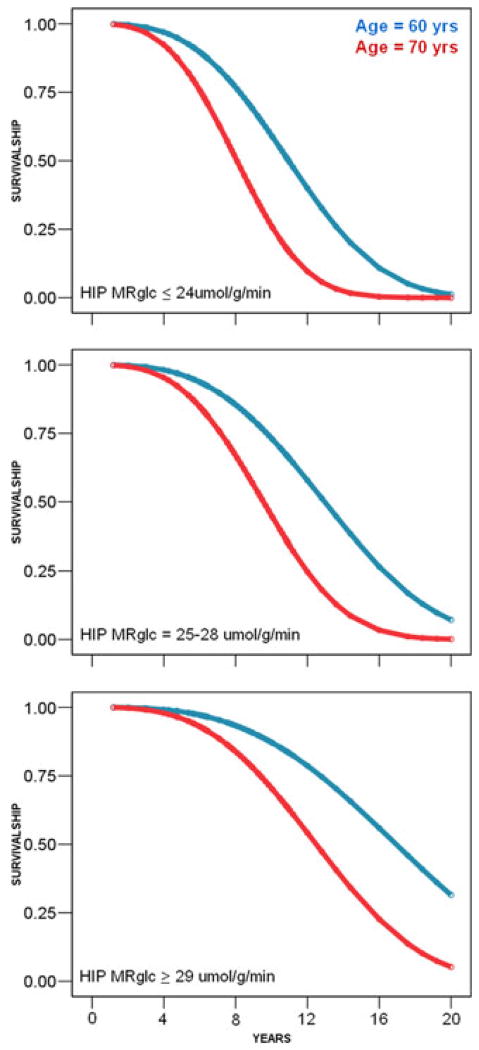
One of the main features of Alzheimer's disease (AD) is the severe reduction of the cerebral metabolic rate for glucose (CMRglc). In vivo imaging using positron emission tomography with 2-[(18)F]fluoro-2-deoxy-D-glucose (FDG-PET) demonstrates consistent and progressive CMRglc reductions in AD patients, the extent and topography of which correlate with symptom severity. Increasing evidence suggests that CMRglc reductions occur at the preclinical stages of AD. CMRglc reductions were observed on FDG-PET before the onset of disease in several groups of at-risk individuals, including patients with mild cognitive impairment (MCI), often a prodrome to AD; presymptomatic individuals carrying mutations responsible for early-onset familial AD; cognitively normal elderly individuals followed for several years until they declined to MCI and eventually to AD; normal, middle-aged individuals who expressed subjective memory complaints and were carriers of the apolipoprotein E epsilon-4 allele, a strong genetic risk factor for late-onset AD. However, the causes of the early metabolic dysfunction forerunning the onset of AD are not known. An increasing body of evidence indicates a deficient or altered energy metabolism that could change the overall oxidative microenvironment for neurons during the pathogenesis and progression of AD, leading to alterations in mitochondrial enzymes and in glucose metabolism in AD brain tissue. The present paper reviews findings that implicate hypometabolism and oxidative stress as crucial players in the initiation and progression of synaptic pathology in AD.
Conflict of interest statement
Conflicts of Interest
The authors declare no conflicts of interest.
Figures

References
-
- Mirra SS, Heyman A, Mckeel D, et al. The consortium to establish a registry for Alzheimer’s disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurol. 1991:479–486. - PubMed
-
- Price JL, Morris JC. Tangles and plaques in nondemented aging and “preclinical” Alzheimer’s disease. Ann Neurol. 1999;45:358–368. - PubMed
-
- Braak H, Braak E. Development of Alzheimer-related neurofibrillary changes in the neocortex inversely recapitulates cortical myelogenesis. Acta Neuropathologica. 1996;92:197–201. - PubMed
-
- Delacourte A, David JP, Sergeant N, et al. The biochemical pathway of neurofibrillary degeneration in aging and Alzheimer’s disease. Neurol. 1999;52:1158–1165. - PubMed
-
- Morris JC, Storandt M, Mckeel DW, et al. Cerebral amyloid deposition and diffuse plaques in “normal” aging: Evidence for presymptomatic and very mild Alzheimer’s disease. Neurol. 1996;46:707–719. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
