

Neural stem cells target intracranial glioma to deliver an oncolytic adenovirus in vivo
- PMID: 19078993
- PMCID: PMC2642530
- DOI: 10.1038/gt.2008.165
Neural stem cells target intracranial glioma to deliver an oncolytic adenovirus in vivo
Abstract
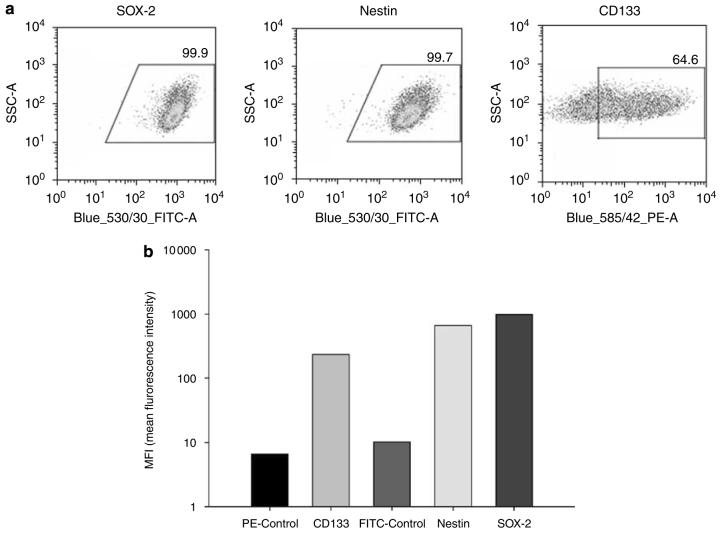
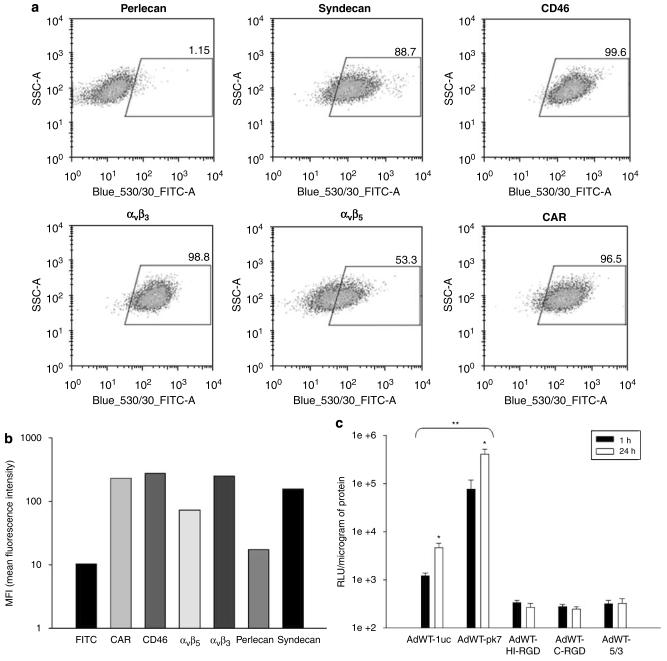
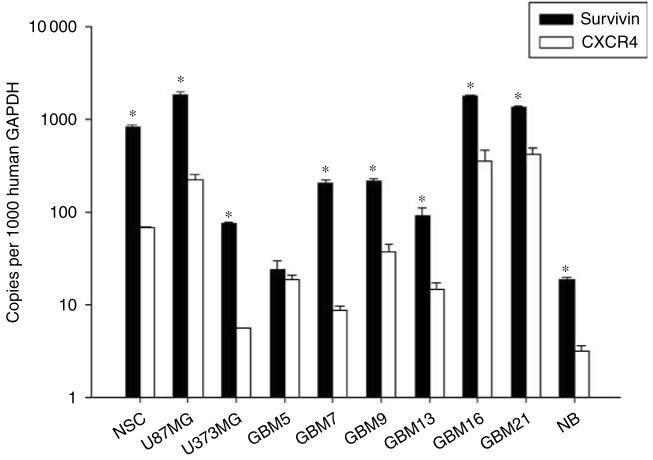
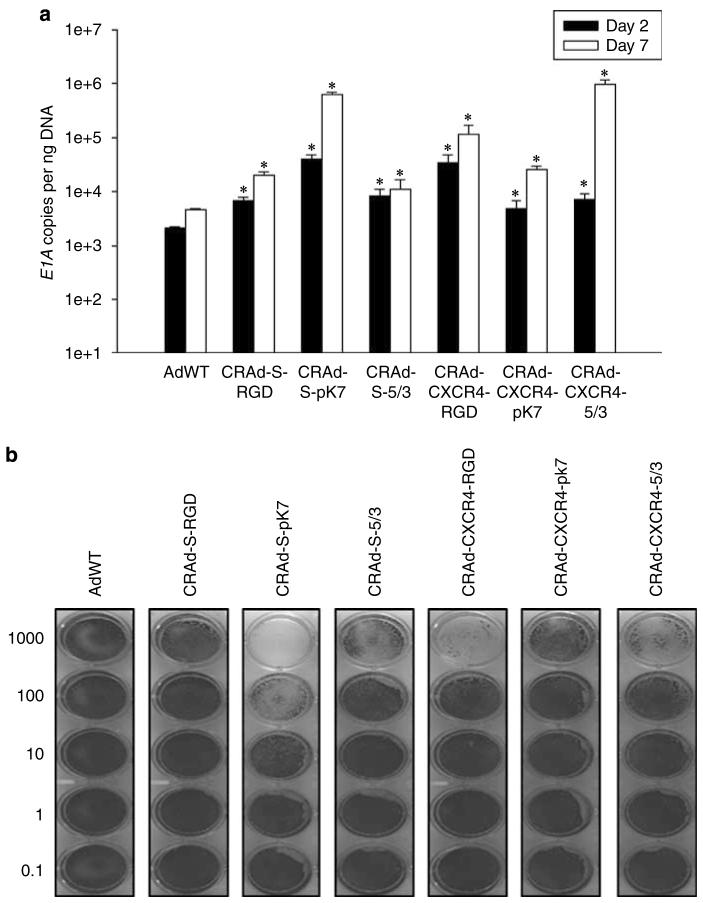
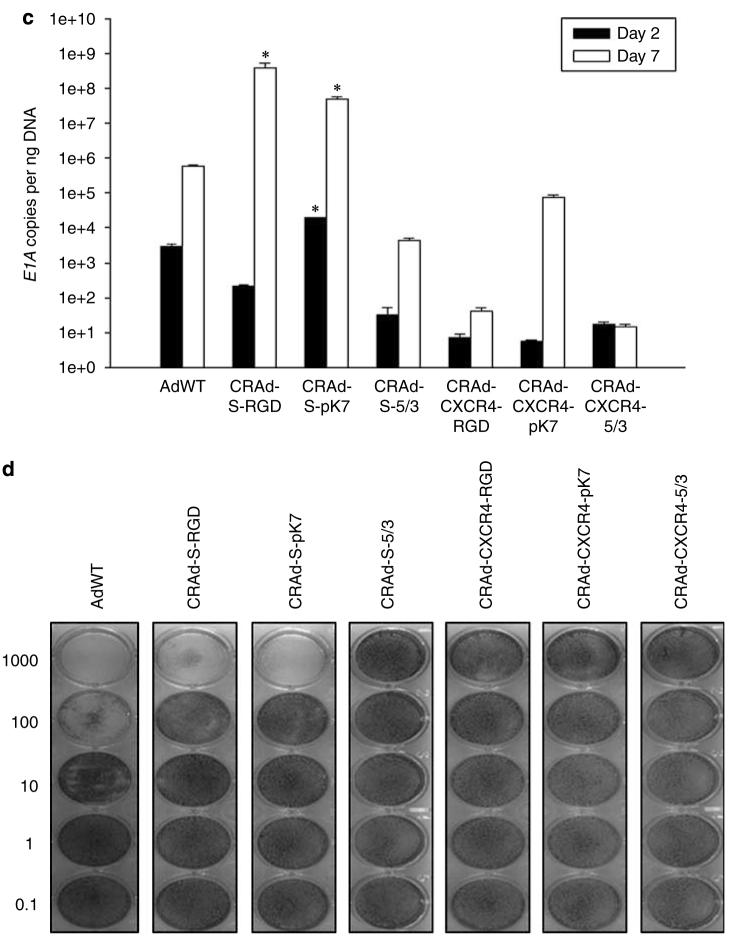
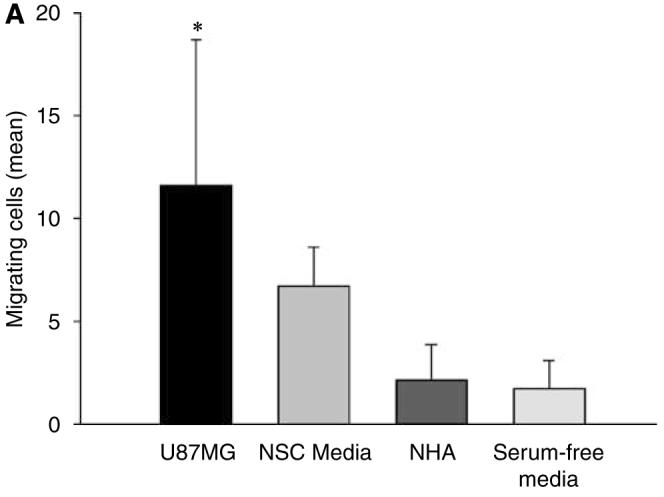
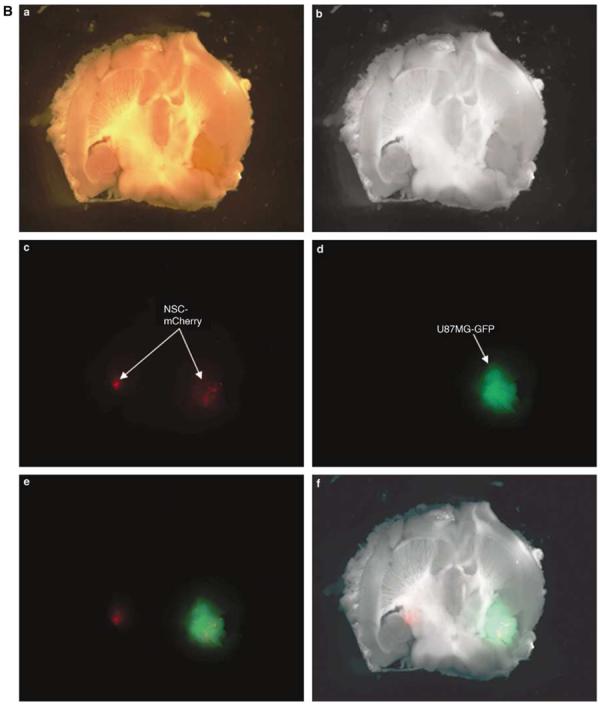
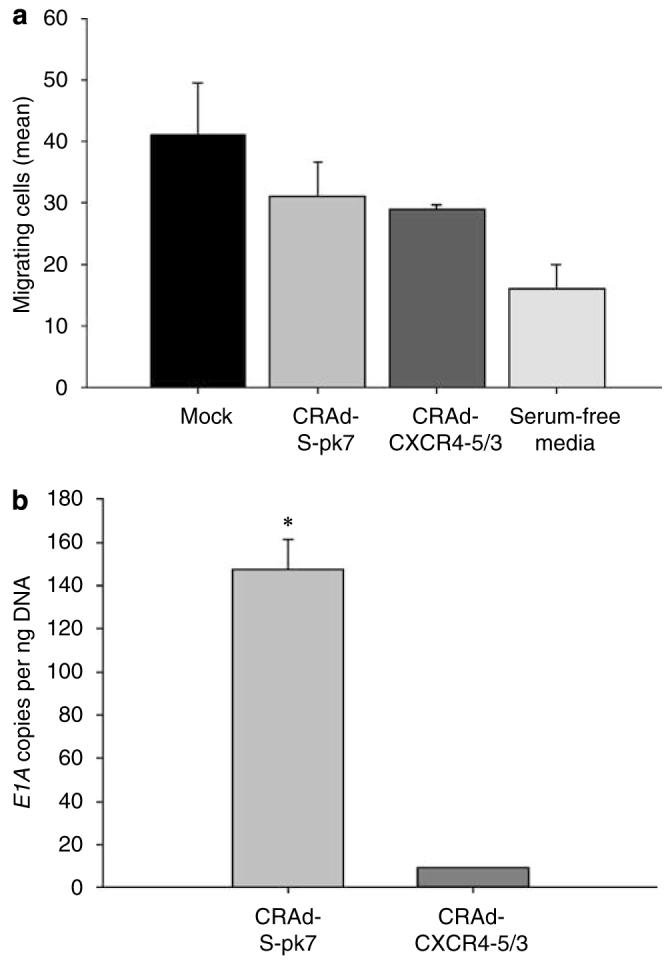
Adenoviral oncolytic virotherapy represents an attractive treatment modality for central nervous system (CNS) neoplasms. However, successful application of virotherapy in clinical trials has been hampered by inadequate distribution of oncolytic vectors. Neural stem cells (NSCs) have been shown as suitable vehicles for gene delivery because they track tumor foci. In this study, we evaluated the capability of NSCs to deliver a conditionally replicating adenovirus (CRAd) to glioma. We examined NSC specificity with respect to viral transduction, migration and capacity to deliver a CRAd to tumor cells. Fluorescence-activated cell sorter (FACS) analysis of NSC shows that these cells express a variety of surface receptors that make them amenable to entry by recombinant adenoviruses. Luciferase assays with replication-deficient vectors possessing a variety of transductional modifications targeted to these receptors confirm these results. Real-time PCR analysis of the replication profiles of different CRAds in NSCs and a representative glioma cell line, U87MG, identified the CRAd-Survivin (S)-pk7 virus as optimal vector for further delivery studies. Using in vitro and in vivo migration studies, we show that NSCs infected with CRAd-S-pk7 virus migrate and preferentially deliver CRAd to U87MG glioma. These results suggest that NSCs mediate an enhanced intratumoral distribution of an oncolytic vector in malignant glioma when compared with virus injection alone.
Figures
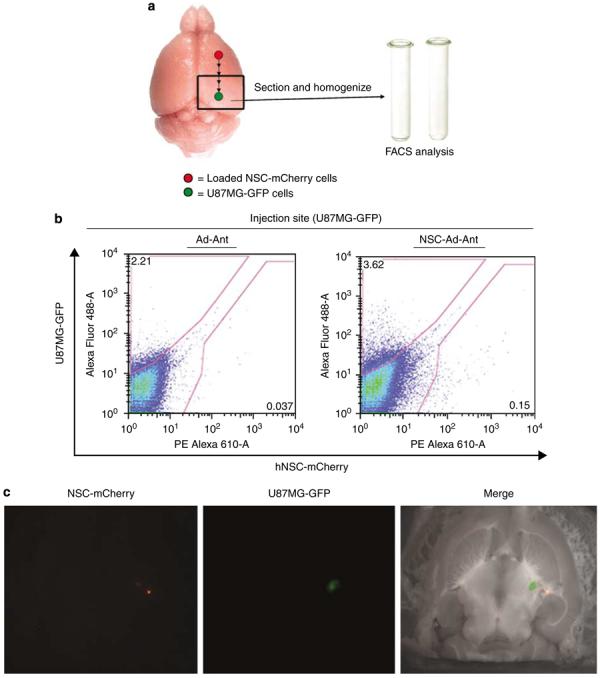
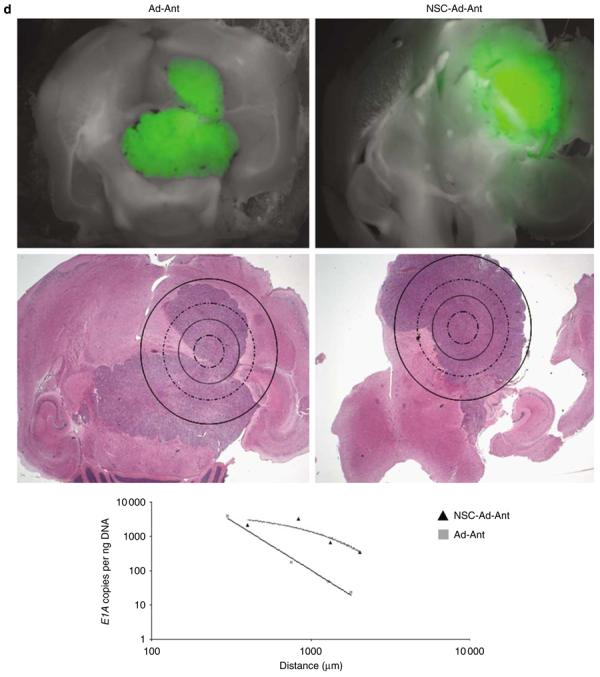
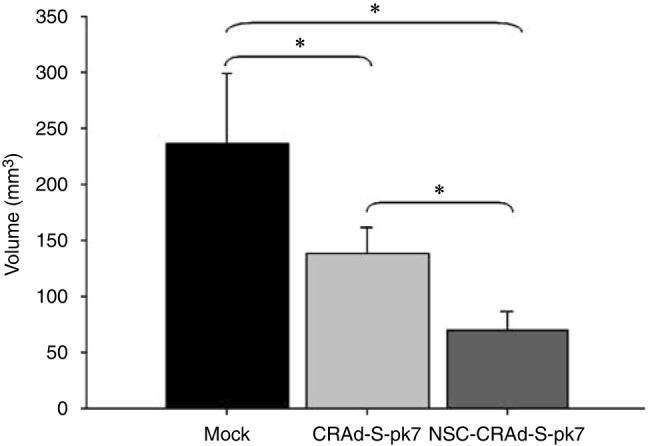
References
-
- Tyler MA, Sonabend AM, Ulasov IV, Lesniak MS. Vector therapies for malignant glioma: shifting the clinical paradigm. Expert Opin Drug Deliv. 2008;5:445–458. - PubMed
-
- Borovjagin AV, Krendelchtchikov A, Ramesh N, Yu DC, Douglas JT, Curiel DT. Complex mosaicism is a novel approach to infectivity enhancement of adenovirus type 5-based vectors. Cancer Gene Ther. 2005;12:475–486. - PubMed
-
- Brouwer E, Havenga MJ, Ophorst O, de Leeuw B, Gijsbers L, Gillissen G, et al. Human adenovirus type 35 vector for gene therapy of brain cancer: improved transduction and bypass of pre-existing anti-vector immunity in cancer patients. Cancer Gene Ther. 2007;14:211–219. - PubMed
-
- Miura Y, Yoshida K, Nishimoto T, Hatanaka K, Ohnami S, Asaka M, et al. Direct selection of targeted adenovirus vectors by random peptide display on the fiber knob. Gene Therapy. 2007;14:1448–1460. - PubMed
-
- Paul CP, Everts M, Dent P, Fisher PB, Ulasov IV, Lesniak MS, et al. Characterization of infectivity of Knob-modified adenoviral vectors in glioma. Cancer Biol Ther. 2008;7:786–793. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
