

Caspases as therapeutic targets in Alzheimer's disease: is it time to "cut" to the chase?
- PMID: 19079646
- PMCID: PMC2583629
Caspases as therapeutic targets in Alzheimer's disease: is it time to "cut" to the chase?
Abstract
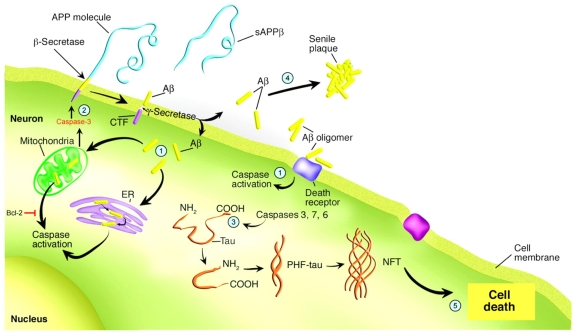
Mounting evidence suggests the involvement of caspases in the disease process associated with Alzheimer's disease (AD). The activation of caspases may be responsible for the neurodegeneration associated with AD and several recent studies have suggested that caspases may also play a role in promoting pathogenic mechanisms underlying this disease. Thus, caspase activation and cleavage of the amyloid precursor protein (APP) and tau may facilitate both the production of beta-amyloid (Abeta) as well as the formation of neurofibrillary tangles (NFTs). Because the activation of caspases in AD may be a proximal event that is not just associated with neurodegeneration, caspases are potential therapeutic targets for the treatment of this disorder. In this review, studies documenting the role of caspases in the AD brain will be discussed. In this context, a discussion of the therapeutic value of targeting caspase inhibition in the treatment of AD will be evaluated including drug targets, delivery and selectivity.
Keywords: Alzheimer’s disease; Apoptosis; Tau; amyloid precursor protein; beta-amyloid; caspase; neurofibrillary tangle.
Figures
References
-
- Golde TE, Dickson D, Hutton M. Filling the gaps in the abeta cascade hypothesis of Alzheimer's disease. Curr Alzheimer Res. 2006;3:421–430. - PubMed
-
- Christensen DD. Alzheimer's disease: progress in the development of anti-amyloid disease-modifying therapies. CNS Spectr. 2007;12:113–116. 119–123. - PubMed
-
- Su JH, Anderson AJ, Cummings BJ, Cotman CW. Immunohistochemical evidence for DNA fragmentation in neurons in the AD brain. Neuroreport. 1994;5:2529–2533. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources