

Mitochondria in neuroplasticity and neurological disorders
- PMID: 19081372
- PMCID: PMC2692277
- DOI: 10.1016/j.neuron.2008.10.010
Mitochondria in neuroplasticity and neurological disorders
Abstract
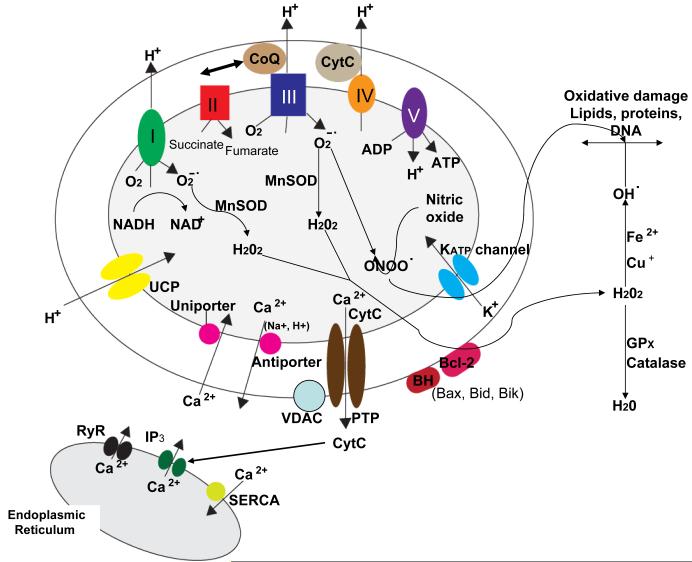
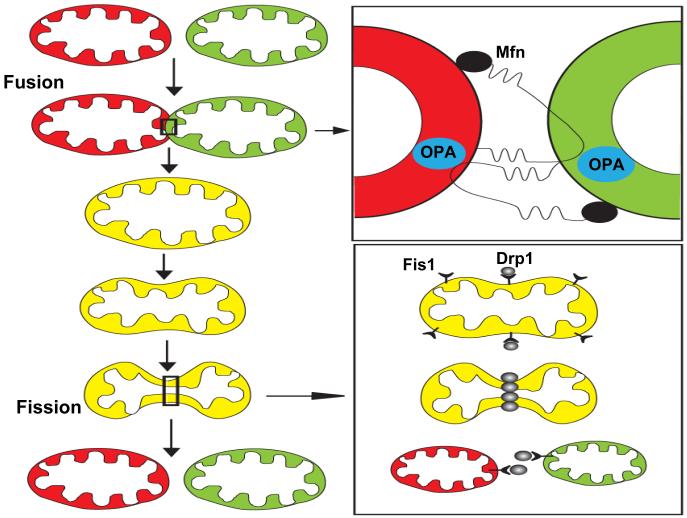
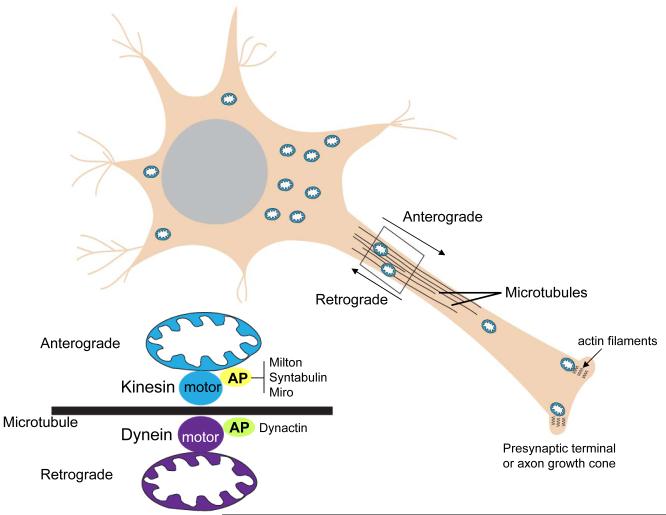
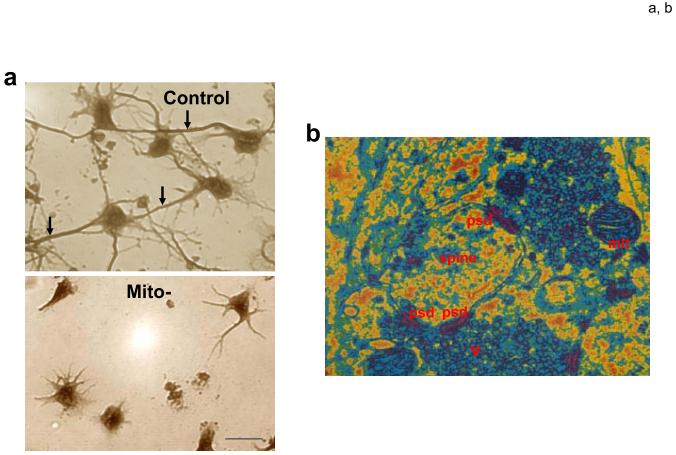
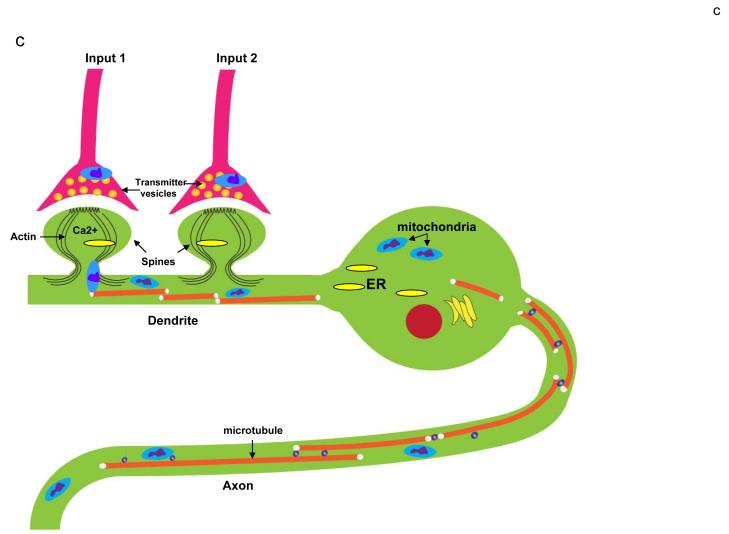
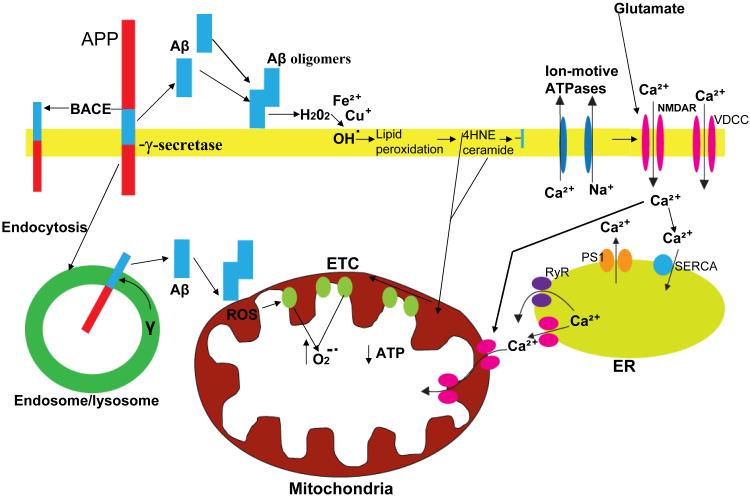
Mitochondrial electron transport generates the ATP that is essential for the excitability and survival of neurons, and the protein phosphorylation reactions that mediate synaptic signaling and related long-term changes in neuronal structure and function. Mitochondria are highly dynamic organelles that divide, fuse, and move purposefully within axons and dendrites. Major functions of mitochondria in neurons include the regulation of Ca(2+) and redox signaling, developmental and synaptic plasticity, and the arbitration of cell survival and death. The importance of mitochondria in neurons is evident in the neurological phenotypes in rare diseases caused by mutations in mitochondrial genes. Mitochondria-mediated oxidative stress, perturbed Ca(2+) homeostasis, and apoptosis may also contribute to the pathogenesis of prominent neurological diseases including Alzheimer's, Parkinson's, and Huntington's diseases; stroke; amyotrophic lateral sclerosis; and psychiatric disorders. Advances in understanding the molecular and cell biology of mitochondria are leading to novel approaches for the prevention and treatment of neurological disorders.
Figures

References
-
- Abeliovich A, Schmitz Y, Fariñas I, Choi-Lundberg D, Ho WH, Castillo PE, Shinsky N, Verdugo JM, Armanini M, Ryan A, et al. Mice lacking alpha-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron. 2000;25:239–252. - PubMed
-
- Arthur PG, Matich GP, Pang WW, Yu DY, Bogoyevitch MA. Necrotic death of neurons following an excitotoxic insult is prevented by a peptide inhibitor of c-jun N-terminal kinase. J. Neurochem. 2007;102:65–76. - PubMed
-
- Arumugam TV, Gleichmann M, Tang SC, Mattson MP. Hormesis/preconditioning mechanisms, the nervous system and aging. Ageing Res. Rev. 2006;5:165–178. - PubMed
-
- Atsumi T. The ultrastructure of intramuscular nerves in amyotrophic lateral sclerosis. Acta Neuropathol. 1981;55:193–198. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
