

Genetic risk factors for variant Creutzfeldt-Jakob disease: a genome-wide association study
- PMID: 19081515
- PMCID: PMC2643048
- DOI: 10.1016/S1474-4422(08)70265-5
Genetic risk factors for variant Creutzfeldt-Jakob disease: a genome-wide association study
Abstract
Background: Human and animal prion diseases are under genetic control, but apart from PRNP (the gene that encodes the prion protein), we understand little about human susceptibility to bovine spongiform encephalopathy (BSE) prions, the causal agent of variant Creutzfeldt-Jakob disease (vCJD).
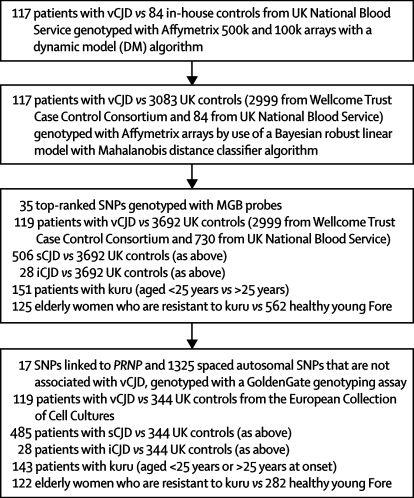
Methods: We did a genome-wide association study of the risk of vCJD and tested for replication of our findings in samples from many categories of human prion disease (929 samples) and control samples from the UK and Papua New Guinea (4254 samples), including controls in the UK who were genotyped by the Wellcome Trust Case Control Consortium. We also did follow-up analyses of the genetic control of the clinical phenotype of prion disease and analysed candidate gene expression in a mouse cellular model of prion infection.
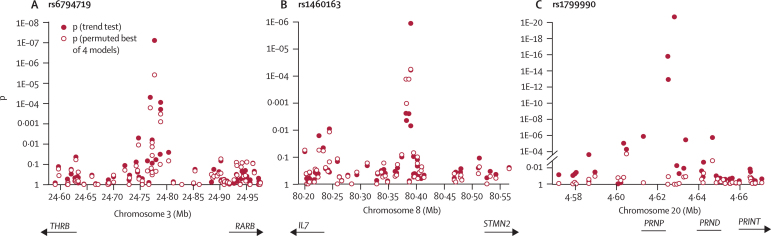
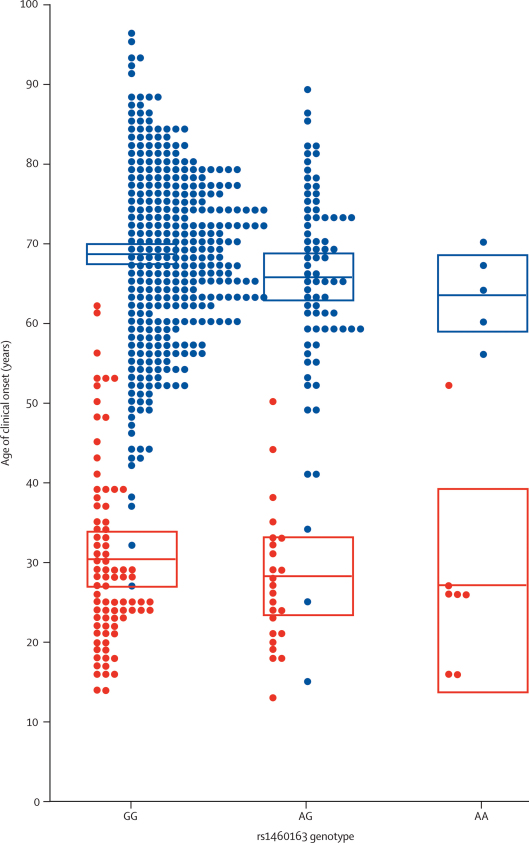
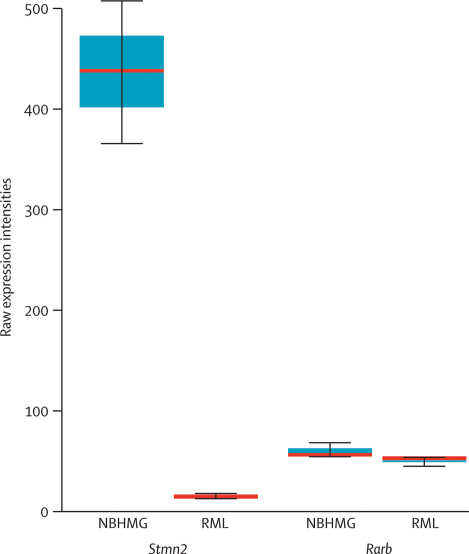
Findings: The PRNP locus was strongly associated with risk across several markers and all categories of prion disease (best single SNP [single nucleotide polymorphism] association in vCJD p=2.5 x 10(-17); best haplotypic association in vCJD p=1 x 10(-24)). Although the main contribution to disease risk was conferred by PRNP polymorphic codon 129, another nearby SNP conferred increased risk of vCJD. In addition to PRNP, one technically validated SNP association upstream of RARB (the gene that encodes retinoic acid receptor beta) had nominal genome-wide significance (p=1.9 x 10(-7)). A similar association was found in a small sample of patients with iatrogenic CJD (p=0.030) but not in patients with sporadic CJD (sCJD) or kuru. In cultured cells, retinoic acid regulates the expression of the prion protein. We found an association with acquired prion disease, including vCJD (p=5.6 x 10(-5)), kuru incubation time (p=0.017), and resistance to kuru (p=2.5 x 10(-4)), in a region upstream of STMN2 (the gene that encodes SCG10). The risk genotype was not associated with sCJD but conferred an earlier age of onset. Furthermore, expression of Stmn2 was reduced 30-fold post-infection in a mouse cellular model of prion disease.
Interpretation: The polymorphic codon 129 of PRNP was the main genetic risk factor for vCJD; however, additional candidate loci have been identified, which justifies functional analyses of these biological pathways in prion disease.
Figures

Comment in
-
Are further genetic factors associated with the risk of developing variant Creutzfeldt-Jakob disease?Lancet Neurol. 2009 Jan;8(1):25-6. doi: 10.1016/S1474-4422(08)70266-7. Lancet Neurol. 2009. PMID: 19081509 No abstract available.
References
-
- Collinge J. Prion diseases of humans and animals: their causes and molecular basis. Annu Rev Neurosci. 2001;24:519–550. - PubMed
-
- Collinge J, Sidle KCL, Meads J, Ironside J, Hill AF. Molecular analysis of prion strain variation and the aetiology of ‘new variant’ CJD. Nature. 1996;383:685–690. - PubMed
-
- Bruce ME, Will RG, Ironside JW. Transmissions to mice indicate that ‘new variant’ CJD is caused by the BSE agent. Nature. 1997;389:498–501. - PubMed
-
- Hill AF, Desbruslais M, Joiner S, Sidle KCL, Gowland I, Collinge J. The same prion strain causes vCJD and BSE. Nature. 1997;389:448–450. - PubMed
-
- Collinge J, Whitfield J, McKintosh E. Kuru in the 21st century—an acquired human prion disease with very long incubation periods. Lancet. 2006;367:2068–2074. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
