

Classic and atypical fibrodysplasia ossificans progressiva (FOP) phenotypes are caused by mutations in the bone morphogenetic protein (BMP) type I receptor ACVR1
- PMID: 19085907
- PMCID: PMC2921861
- DOI: 10.1002/humu.20868
Classic and atypical fibrodysplasia ossificans progressiva (FOP) phenotypes are caused by mutations in the bone morphogenetic protein (BMP) type I receptor ACVR1
Abstract
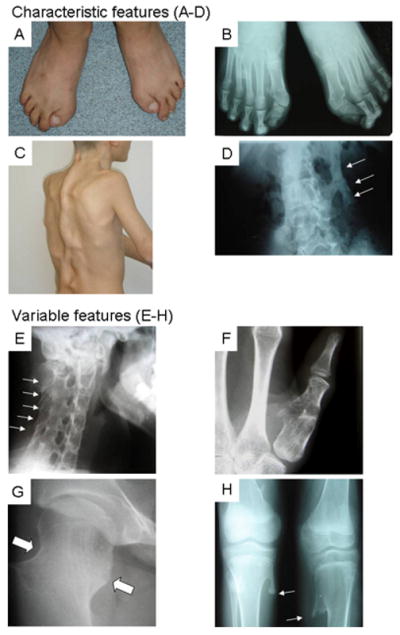
Fibrodysplasia ossificans progressiva (FOP) is an autosomal dominant human disorder of bone formation that causes developmental skeletal defects and extensive debilitating bone formation within soft connective tissues (heterotopic ossification) during childhood. All patients with classic clinical features of FOP (great toe malformations and progressive heterotopic ossification) have previously been found to carry the same heterozygous mutation (c.617G>A; p.R206H) in the glycine and serine residue (GS) activation domain of activin A type I receptor/activin-like kinase 2 (ACVR1/ALK2), a bone morphogenetic protein (BMP) type I receptor. Among patients with FOP-like heterotopic ossification and/or toe malformations, we identified patients with clinical features unusual for FOP. These atypical FOP patients form two classes: FOP-plus (classic defining features of FOP plus one or more atypical features) and FOP variants (major variations in one or both of the two classic defining features of FOP). All patients examined have heterozygous ACVR1 missense mutations in conserved amino acids. While the recurrent c.617G>A; p.R206H mutation was found in all cases of classic FOP and most cases of FOP-plus, novel ACVR1 mutations occur in the FOP variants and two cases of FOP-plus. Protein structure homology modeling predicts that each of the amino acid substitutions activates the ACVR1 protein to enhance receptor signaling. We observed genotype-phenotype correlation between some ACVR1 mutations and the age of onset of heterotopic ossification or on embryonic skeletal development.
2008 Wiley-Liss, Inc.
Figures



References
-
- Ahn J, de la Pena LS, Shore EM, Kaplan FS. Paresis of a bone morphogenetic protein-antagonist response in a genetic disorder of heterotopic skeletogenesis. J Bone Joint Surg (Am) 2003;85A:667–674. - PubMed
-
- Andreev K, Zenkel M, Kruse F, Junemann A, Schlotzer-Schrehardt U. Expression of bone morphogenetic proteins (BMPs), their receptors, and activins in normal and scarred conjunctiva: role of BMP-6 and activin-A in conjunctival scarring? Exp Eye Res. 2006;83:1162–1170. - PubMed
-
- Bakrania P, Efthymiou M, Klein JC, Salt A, Bunyan DJ, Wyatt A, Ponting CP, Mmartin A, Williams S, Lindley V, Gilmore J, Restori M, Robson AG, Neveu MM, Holder GE, Collin JRO, Robinson DO, Fardon P, Johansen-Berg H, Gerrelli D, Ragge NK. Mutations in BMP4 cause eye, brain, and digit developmental anomalies: Overlap between the BMP4 and Hedgehog signaling pathways. Am J Hum Genet. 2008;82:304–319. - PMC - PubMed
-
- Baur ST, Mai JJ, Dymecki SM. Combinatorial signaling through BMP receptor IB and GDF5: shaping of the distal mouse limb and the genetics of distal limb diversity. Development. 2000;127:605–619. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
