

Mitochondrial dysfunction in mut methylmalonic acidemia
- PMID: 19088183
- PMCID: PMC2660647
- DOI: 10.1096/fj.08-121848
Mitochondrial dysfunction in mut methylmalonic acidemia
Abstract
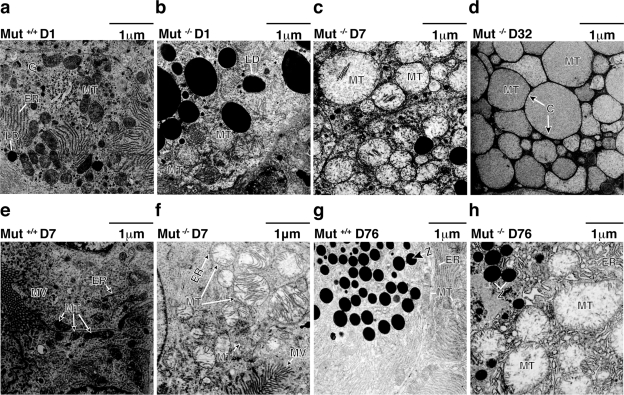
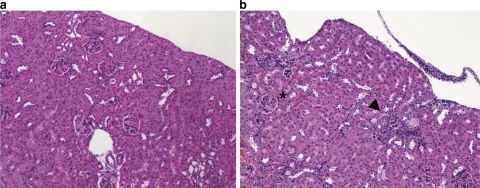
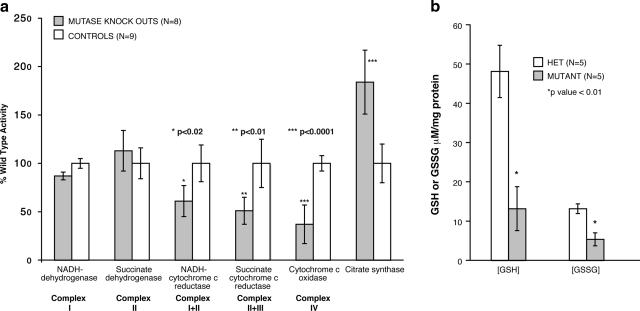
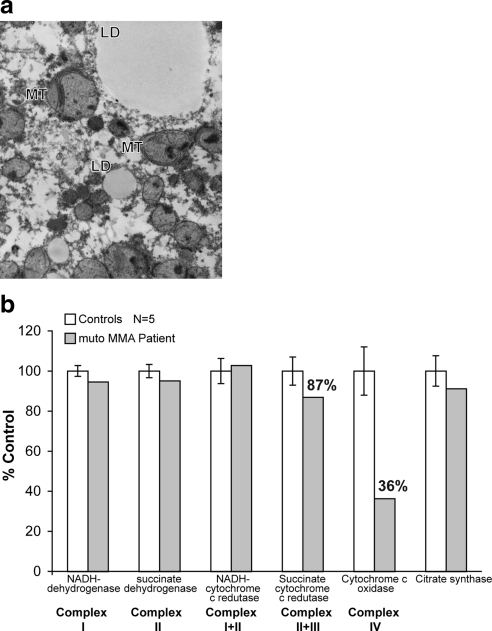
Methylmalonic acidemia is an autosomal recessive inborn error of metabolism caused by defective activity of methylmalonyl-CoA mutase (MUT) that exhibits multiorgan system pathology. To examine whether mitochondrial dysfunction is a feature of this organic acidemia, a background-modified Mut-knockout mouse model was constructed and used to examine mitochondrial ultrastructure and respiratory chain function in the tissues that manifest pathology in humans. In parallel, the liver from a patient with mut methylmalonic acidemia was studied in a similar fashion. Megamitochondria formed early in life in the hepatocytes of the Mut(-/-) animals and progressively enlarged. Liver extracts prepared from the mutants at multiple time points displayed respiratory chain dysfunction, with diminished cytochrome c oxidase activity and reduced intracellular glutathione compared to control littermates. Over time, the exocrine pancreas and proximal tubules of the kidney also exhibited megamitochondria, and older mutant mice eventually developed tubulointerstitial renal disease. The patient liver displayed similar morphological and enzymatic findings as observed in the murine tissues. These murine and human studies establish that megamitochondria formation with respiratory chain dysfunction occur in a tissue-specific fashion in methylmalonic acidemia and suggest treatment approaches based on improving mitochondrial function and ameliorating the effects of oxidative stress.
Figures
References
-
- Fenton W A, Gravel R A, Rosenblatt D S. Disorders of propionate and methylmalonate metabolism. Scriver C R, Beaudet A L, Sly W S, Valle D, Childs B, Kinzler K W, Vogelstein B, editors. New York: McGraw-Hill; The Metabolic and Molecular Bases of Inherited Disease. 2001:2165–2192.
-
- Fenton W A, Rosenblatt D S. Inherited disorders of folate and cobalamin transport and metabolism. Scriver C R, Beaudet A L, Sly W S, Valle D, Childs B, Kinzler K W, Vogelstein B, editors. New York: McGraw-Hill; The Metabolic and Molecular Bases of Inherited Disease. 2001:3897–3933.
-
- Matsui S M, Mahoney M J, Rosenberg L E. The natural history of the inherited methylmalonic acidemias. N Engl J Med. 1983;308:857–861. - PubMed
-
- Baumgarter E R, Viardot C. Long-term follow-up of 77 patients with isolated methylmalonic acidaemia. J Inherit Metab Dis. 1995;18:138–142. - PubMed
-
- Van der Meer S B, Poggi F, Spada M, Bonnefont J P, Ogier H, Hubert P, Depondt E, Rapoport D, Rabier D, Charpentier C, Parvy P, Kamoun P, Saudubray J M. Clinical outcome of long-term management of patients with vitamin B12-unresponsive methylmalonic acidemia. J Pediatr. 1994;125:903–908. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
